 2021, Vol. 32
2021, Vol. 32 

b Department of Chemistry, Xihua University, Chengdu 610039, China
Fused indolizidines and quinolizidines are found in a variety of natural products as well as in pharmacologically important compounds and possess a wide spectrum of biological activities [1-5] (Fig. 1).

|
Download:
|
| Fig. 1. Related natural products and drugs containing fused indolizidine or quinolizidine structures. | |
Inspired by the important value of these compounds in modern pharmacology [6, 7], scholars have devoted significant efforts to their construction [8-15]. Among them, Bischler–Napieralski reaction (B–N reaction) [16, 17] offers a very important strategy (Scheme 1a). However, harsh reaction conditions or highly corrosive reagents such as POCl3 and P2O5 are involved in traditional B–N reaction. Moreover, to obtain the target fused ring, multiple steps of reactions are imperative, leading to reduced yields and tedious operation due to the indispensable isolation and purification of intermediates [8, 9]. Although some other existing methods are efficient, they also suffer from some inevitable drawbacks, such as using hazardous reagents [10], expensive metal catalysts [11, 12] or commercially unavailable catalysts or substrates [13, 9-15].

|
Download:
|
| Scheme 1. Comparison of previous works and this work. | |
In recent decades, amide activation has become an emerging tool for chemoselective synthesis [18, 19]. Researchers such as Movassaghi [20, 21] and Huang [22] have conducted elegant works in this field.
In 2008, Movassaghi's group reported a method to synthesize isoquinoline and β-carboline derivatives through a mild B–N reaction by using trifluoromethanesulfonic anhydride (Tf2O) to activate amide [20]. But unsubstituted indole derivatives were not suitable for this method to synthesize correspondingβ-carboline derivatives, which limited its application scope to a certain extent. And the synthesis of structurally more complicated fused indolizidines and quinolizidines was also not involved (Scheme 1b).
As part of our program to develop practical method for the construction of pharmaceutical products [23, 24], herein, a one-pot tandem route [25-27] based on amide activation to fused indolizidines and quinolizidines is disclosed (Scheme 1c).
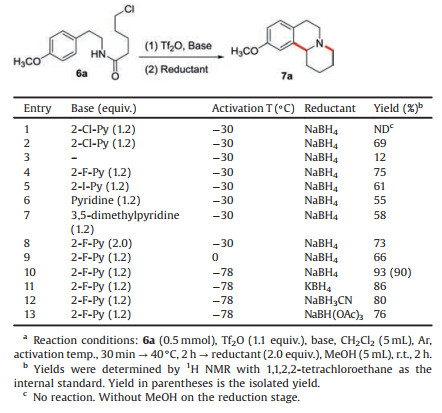
5-Chloro-N-(4-methoxyphenethyl)pentanamide (6a) was selected as the starting material for optimization of reaction conditions. The intermediate dihydroisoquinoline other than the desired tricyclic product (7a) was detected under the initial reaction conditions (Table 1, entry 1). This finding suggested that the activation of amide and B–N reaction went well, but reduction following cyclization did not occur. This phenomenon might be due to the poor solubility of the reductant NaBH4 in the aprotic solvent CH2Cl2. The addition of methanol solved the problem satisfactorily, giving the target product in 69% yield (Table 1, entry 2). We then screened potential bases (Table 1, entries 3–7), and 2-fluoropyridine (2-F-Py) was found to be the most efficient (Table 1, entry 4). Nevertheless, increasing the amount of 2-F-Py did not contribute to the improvement 9 and 10 of yield (Table 1, entry 8). When base was absent, the reaction also occurred in spite of reduced yield (Table 1, entry 3). The activation temperature was subsequently investigated (Table 1, entries 9–10). With the activation temperature decreased to −78 ℃, the product was obtained in 93% yield (Table 1, entry 10). Other different reductants including KBH4, NaBH3CN, and NaBH(OAc)3 were then tested, but the outcomes were inferior to NaBH4.
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
{kind=link}
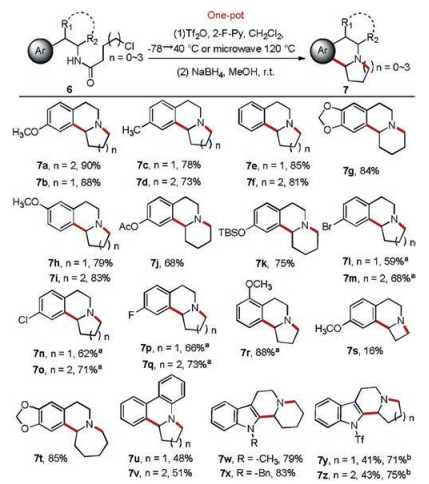
We examined the substrate scope under the optimized conditions. Substrates with electron-donating substitutions or without any substitution in the aromatic nucleus all reacted smoothly to generate corresponding fused indolizidines and quinolizidines in good to excellent yields (Scheme 2, 7a–7k). The reaction system was applied to substrates containing somewhat electron-withdrawing groups. However, no desired tricyclic products were detected. This was probably because the reduced electron density on the aromatic ring led to difficulty in the occurrence of the B–N reaction. Nevertheless, with the aid of microwave, tricyclic compounds with somewhat electron-withdrawing groups such as F-, Cl- and Br- were obtained in moderate to good yields (Scheme 2, 7l–7q). The substitution position also affected the reaction. The reaction with the substitute group in para and meta positions proceeded smoothly under the regular condition, whereas that with ortho substitution reacted only under microwave (Scheme 2, 7r). We further investigated the impact of ring size on the reaction. The results showed that seven-membered fused ring was easily formed, while four-membered fused ring was more difficult to obtain (Scheme 2, 7s–7t). In addition, fused dihydrophenanthridine derivatives were obtained in moderate yields using o-arylaniline-derived amides as substrates through the Morgan–Walls reaction [28] (Scheme 2, 7u–7v).

|
Download:
|
| Scheme 2. Scope of this method. Uniform reaction conditions unless otherwise noted: Compound 6 (0.5 mmol), Tf2O (1.1 equiv.), 2-F-Py (1.2 equiv.), CH2Cl2 (5 mL), Ar, −78 ℃, 30 min → 40 ℃, 2h → MeOH (5 mL), NaBH4 (2.0 equiv.), r.t., 2 h.a −78 ℃, 30 min → microwave, 120 ℃, 10 min → r.t., 2 h.b Tf2O (2.2 equiv.). | |
{kind=link}
Indole derivated fused indolizidines and quinolizidines were also synthesized. Trypta mine-derived amides with indole nitrogen protected by methyl or benzyl were converted into corresponding products in good yields (Scheme 2, 7w–7x). It was worth noting that unsubstituted indole derivatives led to rapid indolyl nitrogen sulfonylation by Tf2O, and these compounds were unsuitable for Movassaghi's method [20] to be activated and underwent B–N reaction. Under our reaction condition, indole derivatives without any substitution were converted into fused tetrahydro-β-carbolines with sulfonylations on indolyl nitrogen. The relatively low yields might be caused by insufficient Tf2O which was consumed by indolyl nitrogen sulfonylation. When Tf2O was adjusted to 2.2 equiv., the yields were improved obviously (Scheme 2, 7y–7z).
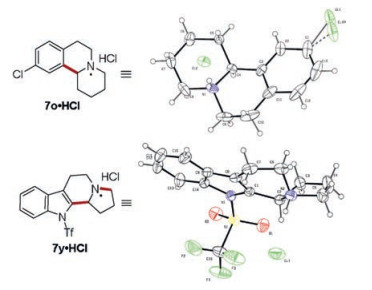
Compounds 7o and 7y were converted into hydrochlorides, and their structures were unequivocally confirmed by X-ray crystal structure deter mination (Fig. 2).

|
Download:
|
| Fig. 2. X-ray crystal structure of 7o·HCl and 7y·HCl. | |
{kind=link}
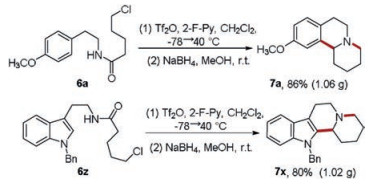
Additionally, gram-scale syntheses of typical fused quinolizidines were also realized through the protocol (Scheme 3).

|
Download:
|
| Scheme 3. Gram-scale synthesis. | |
{kind=link}
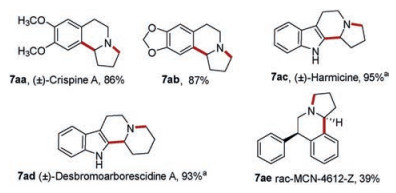
Our method was further applied to synthesize natural products or bioactive compounds. (±)-Crispine A (7aa, Scheme 4) shows cytotoxic activity against HeLa human cancer cell lines [29]. It was synthesized smoothly by using our one-pot method in good yield. A number of methods have been reported to synthesize (±)-crispine A, which undergoes multiple reaction steps in different ways [30-33], however, our protocol is one of the most efficient. Compound 7ab (Scheme 4) is also a pharmacological active substance that displays potent antagonist activity at α4β2 nicotinic acetylcholine receptors (nAChR), a ligand-gated ion channel involved in a broad range of psychiatric and neurodegenerative disorders. In the reported method to synthesize compound 7ab, highly corrosive POCl3 and multi-step microwave-assisted reaction with the temperature as high as 150 ℃ were necessary [34]. Herein, synthesis was conducted in milder condition and more convenient method.

|
Download:
|
| 4. Application of this method to the synthesis of natural products and bioactive compounds. Uniform reaction conditions unless otherwise noted: Compound 6 (0.5 mmol), Tf2O (1.1 equiv.), 2-F-Py (1.2 equiv.), CH2Cl2 (5 mL), Ar, −78 ℃, 30 min → 40 ℃, 2h → MeOH (5 mL), NaBH4 (2.0 equiv.), r.t., 2 h.a Compound 7y or 7z (0.2 mmol), NaOH (4.0 mmol), H2O (4 mL), MeOH (16 mL), reflux, 2 h. | |
{kind=link}
(±)-Harmicine (7ac, Scheme 4) exhibits strong antileishmania activity [35] and (±)-desbromoarborescidine A (7ad, Scheme 4) was an alkaloid discovered from the New Guinea tree Dracontomelum mangiferum [12]. These compounds were easily obtained from the detrifluoromethylsulfonylation of 7y and 7z in NaOH solution (Scheme 4).
MCN-4612-Z (7ae, Scheme 4) is a potent uptake inhibitor for norepinephrine (NE), dopa mine (DA) and serotonin (5-HT), which was developed as antidepressant drugs in 1980s [36]. It was synthesized using our protocol in 39% yield. And its diastereoisomer (7af, MCN-4612-E, Supporting information) [37] was also isolated in the same reaction in 45% yield.
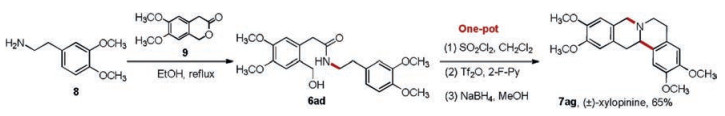
Furthermore, a new strategy was designed to synthesize protoberberine (±)-xylopinine (7ag, Scheme 5). (±)-Xylopinine is an alkaloid found in the stem of Xylopia laevigata (Annonaceae), which exhibits strong in vitro cytotoxic activity toward various tumor cell lines [38]. The amide substrate (6ad, Scheme 5) was synthesized from 3, 4-dimethoxyphenylethyla mine (8, Scheme 5) and 6, 7-dimethoxyisochroman-3-one (9, Scheme 5), which was then subjected to chlorination, B–N reaction, reduction of Schiff base intermediate, and intramolecular cyclization in one-pot to offer (±)-xylopinine as a racemate in 65% total yield. Among the reported synthesis strategies for tetrahydroberberines [39, 40, 41], our method is the one of the most efficient.

|
Download:
|
| Scheme 5. Total synthesis of protoberberine (±)-xylopinine. | |
{kind=link}
A plausible mechanism is illustrated in Scheme 6 taking compound7g as an example. Amide substrate (6g, Scheme 6) was firstly activated by Tf2O to obtain a nitrilium ion [42, 43] (10, Scheme 6), which underwent Bischler–Napieralski reaction to obtain cyclic i minium ion (11, Scheme 6). The iminium ion 11 was isolated and its structure was confirmed by 1H NMR, 13C NMR and HRMS. 11 was subjected to reduction of the C=N bond (12, Scheme 6) followed by intramolecular nucleophilic substitution to obtain polycyclic product 7g.

|
Download:
|
| Scheme 6. The proposed mechanism. | |
{kind=link}
In summary, we propose a facile tandem protocol to construct fused indolizidines and quinolizidines. The protocol is efficient because it integrates three step reactions in one-pot and does not require isolation and purification of any intermediate. The protocol can be easily scaled up to gram-scale. Taking advantage of the mild reaction conditions and functional group tolerance, the protocol was successfully applied to synthesize natural products such as (±)-crispine A, (±)-desbromoarborescidine A, (±)-harmicine, (±)-xylopinine and other bioactive substances.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (No. 81973189), Sichuan Outstanding Young Scientific and Technological Talents Project (No. 2020JDJQ0054), Sichuan Science and Technology Program (No. 19ZDYF0890), Scientific Research Foundation of the Education Department of Sichuan Province (No. 18TD0023) and Xihua University Talents Project (No. Z202030).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2021.01.018.
| [1] |
L. Wu, H. Ling, L. Li, L. Jiang, M. He, J. Pharm. Pharmacol. 59 (2007) 695-701. |
| [2] |
A. Padwa, M.D. Danca, Org. Lett. 4 (2002) 715-717. DOI:10.1021/ol017193v |
| [3] |
A.M. Peckham, J.A. Nicewonder, J. Pharm. Pract. 32 (2019) 450-457. DOI:10.1177/0897190018756512 |
| [4] |
S.(Bas)A.M.W. van den Broek, J.G.H. Lemmers, F.L. van Delft, F.P.J.T. Rutjes, Org. Biomol. Chem. 10 (2012) 945-951. DOI:10.1039/C1OB06539D |
| [5] |
J. Wang, X. Lv, J. Xu, et al., J. Med. Chem. 188 (2020) 111976. DOI:10.1016/j.ejmech.2019.111976 |
| [6] |
H. Wang, Q. Dong, Q. Xie, P. Tang, Chin. Chem. Lett. 31 (2020) 685-688. DOI:10.1016/j.cclet.2019.08.033 |
| [7] |
Y. Zhang, K. Xie, A. Liu, et al., Chin. Chem. Lett. 30 (2019) 443-446. DOI:10.1016/j.cclet.2018.05.010 |
| [8] |
C. Weimar, S. Von Angerer, W. Wiegrebe, Arch. Pharm. (Weinheim) 18 (1991) 509-518. |
| [9] |
B. Hoefgen, M. Decker, P. Mohr, et al., J. Med. Chem. 49 (2006) 760-769. DOI:10.1021/jm050846j |
| [10] |
W.H. Pearson, W.K. Fang, J. Org. Chem. 65 (2000) 7158-7174. DOI:10.1021/jo0011383 |
| [11] |
S. Zhou, R. Tong, Chem. Eur. J. 22 (2016) 7084-7089. DOI:10.1002/chem.201601245 |
| [12] |
E. Park, C.H. Cheon, Org. Biomol. Chem. 15 (2017) 10265-10275. DOI:10.1039/C7OB02691A |
| [13] |
K. Diker, K. El Biach, J. Lévy, J. Nat. Prod. 3 (1997) 791-793. |
| [14] |
N. Meyer, T. Opatz, European J. Org. Chem. 2006 (2006) 3997-4002. DOI:10.1002/ejoc.200600314 |
| [15] |
Y. Zhang, R.P. Hsung, X. Zhang, et al., Org. Lett. 7 (2005) 1047-1050. DOI:10.1021/ol0473391 |
| [16] |
A. Bischler, B. Napieralski, Ber (1933) 1903-1908. |
| [17] |
Z. Wang, Z. Yu, Y. Yao, et al., Chin. Chem. Lett. 30 (2019) 1541-1544. DOI:10.1016/j.cclet.2019.07.001 |
| [18] |
D. Kaiser, N. Maulide, J. Org. Chem. 81 (2016) 4421-4428. DOI:10.1021/acs.joc.6b00675 |
| [19] |
D. Kaiser, A. Bauer, M. Lemmerer, N. Maulide, Chem. Soc. Rev. 47 (2018) 7899-7925. DOI:10.1039/C8CS00335A |
| [20] |
M. Movassaghi, M.D. Hill, Org. Lett. 10 (2008) 3485-3488. DOI:10.1021/ol801264u |
| [21] |
K.L. White, M. Movassaghi, J. Am. Chem. Soc. 138 (2016) 11383-11389. DOI:10.1021/jacs.6b07623 |
| [22] |
H.H. Huo, H.K. Zhang, X.E. Xia, P.Q. Huang, Org. Lett. 14 (2012) 4834-4837. DOI:10.1021/ol302165d |
| [23] |
X. Ran, Y. Long, S. Yang, et al., Org. Chem. Front. 7 (2020) 472-481. DOI:10.1039/C9QO01321K |
| [24] |
L. Yang, X. Ma, C. Yuan, et al., Eur. J. Med. Chem. 134 (2017) 230-241. DOI:10.1016/j.ejmech.2017.04.010 |
| [25] |
Z. Cao, Q. Zhu, Y.W. Lin, W.M. He, Chin. Chem. Lett. 30 (2019) 2132-2138. DOI:10.1016/j.cclet.2019.09.041 |
| [26] |
Q.W. Gui, F. Teng, S.N. Ying, et al., Chin. Chem. Lett. 31 (2020) 3241-3244. DOI:10.1016/j.cclet.2020.07.017 |
| [27] |
W.B. He, L.Q. Gao, X.J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1895-1898. DOI:10.1016/j.cclet.2020.02.011 |
| [28] |
G.T. Morgan, L.P. Walls, J. Chem. Soc. (1905) 2447-2456. |
| [29] |
K.R. Bailey, A.J. Ellis, R. Reiss, T.J. Snape, N.J. Turner, Chem. Commun. 44 (2007) 3640-3642. |
| [30] |
G. Pandey, S.K. Tiwari, B. Singh, Tetrahedron Lett. 57 (2016) 4480-4483. DOI:10.1016/j.tetlet.2016.08.073 |
| [31] |
S. Saha, Ch.V.R. Reddy, B. Patro, Tetrahedron Lett. 52 (2011) 4014-4016. DOI:10.1016/j.tetlet.2011.05.117 |
| [32] |
H.J. Knölker, S. Agarwal, Tetrahedron Lett. 46 (2005) 1173-1175. DOI:10.1016/j.tetlet.2004.12.066 |
| [33] |
S. Agarwal, O. Kataeva, U. Schmidt, H.J. Knölker, RSC Adv. 3 (2013) 1089-1096. DOI:10.1039/C2RA22823H |
| [34] |
F. Crestey, A.A. Jensen, C. Soerensen, et al., J. Med. Chem. 61 (2018) 1719-1729. DOI:10.1021/acs.jmedchem.7b01895 |
| [35] |
T.S. Kam, K.M. Sim, Phytochemistry 47 (1998) 145-147. DOI:10.1016/S0031-9422(97)00513-X |
| [36] |
B.E. Maryanoff, D.F. McComsey, M.J. Costanzo, et al., J. Med. Chem. 27 (1984) 943-946. DOI:10.1021/jm00374a001 |
| [37] |
S.J. Gharpure, D.S. Vishwakarma, R.K. Patel, Chem. Commun. 55 (2019) 6858-6861. DOI:10.1039/C9CC03127H |
| [38] |
L.R.A. Menezes, C.O. D'Sousa Costa, A.C.B.D.C. Rodrigues, et al., Molecules 21 (2016) 1-10. |
| [39] |
P.W. Jeffs, The protoberberine alkaloids, in: R.H.F. Manske (Ed. ), The Alkaloids: Chemistry and Physiology, Academic Press, 1967, pp. 41-115.
|
| [40] |
S. Zhou, R. Tong, Org. Lett. 19 (2017) 1594-1597. DOI:10.1021/acs.orglett.7b00414 |
| [41] |
B. Horst, M.J. Wanner, S.I. Jørgensen, H. Hiemstra, van Maarseveen J.H., J. Org. Chem. 83 (2018) 15110-15117. DOI:10.1021/acs.joc.8b02378 |
| [42] |
A.B. Charette, M. Grenon, Can. J. Chem. 79 (2001) 1694-1703. DOI:10.1139/v01-150 |
| [43] |
J.L. Ye, Y.N. Zhu, H. Geng, P.Q. Huang, Sci. China Chem. 61 (2018) 687-694. DOI:10.1007/s11426-017-9160-1 |