 2021, Vol. 32
2021, Vol. 32 

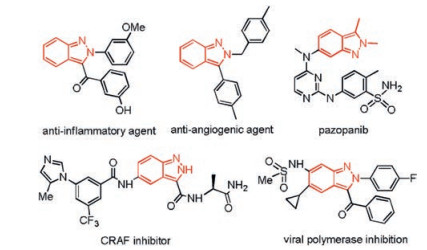
Indazole skeleton is favored by medicinal chemists for their widespread presence in natural products, pharmaceuticals and bioactive compounds [1]. Particularly, 2H-indazoles have recently emerged as promising viral polymerase inhibition, anticancer agents, and anti-inflammatory activities (Fig. 1) [2]. Consequently, numerous efforts have been devoted to construct such a skeletal motif [3]. However, the known procedures toward 2H-indazoles often suffer from multistep synthetic precursors, harsh reaction conditions and limited substrate scope. Therefore, novel efficient strategies to access such a skeleton are highly desirable.

|
Download:
|
| Fig. 1. Selected examples illustrating the importance of 2H-indazole moiety.a | |
Transition-metal-catalyzed directly inter molecular cyclization via C–H bond activation has been established as a straightforward method to construct heterocycles [4]. Among which, the rhodium complex as a catalyst with excellent activity plays a vital role in organic synthesis [5]. As the same time, the azobenzene derivatives are the most classical and conventional compounds in C-H activation/annulation under metal catalysts. Since 2013, the C-H functionalization of azobenzenes as a directing group for constructing various bioactive molecules have been reported using Pd, Co, Rh or Re as catalysis [6]. In these reactions, sp2- or sp-carbons, such as aldehydes, alkynes, alkenes, sulfoxonium ylides and diazo compounds, were usually used as the key synthons for the C-H bond activation/cyclization process. However, sp3-carbon synthons have been rarely reported. To solve this limitation, Glorius's group [7] reported a pioneering example of rhodium(Ⅲ)-catalyzed [4+2] cyclization to synthesize C3-monosubstituted N-heterocycle based on C(sp3) electrophiles of α-Cl/MsO/TsO substituted ketones, complementing the previously reported mode of π-bond reaction. Subsequently, Li and co-workers [8] disclosed the synthesis of C-H/N-H functionalized isoquinolines catalyzed by rhodium(Ⅲ) starting from α-Cl/MsO/TsO ketones. In these procedures, α-halos or pseudohalogens are usually used as C2 synthons to construct these useful frameworks [7-9]. Very recently, Liu and colleagues [10] pioneered the coupling of rhodium(Ⅲ)-catalyzed aromatic C(sp2)-H with α-Cl ketones as a C1 synthon [11] for the synthesis of 3-acylindoles. To build versatile heterocycles as well as with our continuing interest in the clean transition-metal-catalyzed C-H bond functionalization [12], we embark on rhodium(Ⅲ) catalyzed [4+1] cyclization of azo compounds with α-Cl ketones for the synthesis of 3-acyl-2H-indazoles. Cheap and easily available azobenzenes and α-Cl ketones allow the reaction to proceed smoothly under the mild reaction conditions with excellent yields and large functional groups tolerance. Additional, the azo group not only participates as a directing group but also as key component in the core skeleton.
We initially started our investigation by employing azobenzene 1a and α-Cl acetophenone 2a as the model substrates to screen various reaction parameters (Table 1). The desired 2-phenyl-3-benzoyl-2H-indazol 3aa was obtained in an isolated 9.0% yield when [Cp*RhCl2]2 (5.0 mol%)/AgSbF6 (20 mol%) was used as a catalyst and Cu(OAc)2 (0.50 equiv.) as an additive in MeOH at 110 ℃ under air atmosphere (entry 1). Meanwhile, no desired conversion was observed using other acetate salts, including Zn(OAc)2, NaOAc (entries 2 and 3). When CuCO3·Cu(OH)2 (1.0 equiv.) was used as a co-additive, the yield of 3aa was significantly improved to 46% (entry 4). Other co-additives, such as HOAc (36%), K2S2O8 (N.R) and LiCO3 (N.R), were not as effective as CuCO3·Cu(OH)2 (entries 5–7). Subsequently, the effect of solvent was evaluated and 1, 2-dichloroethane (DCE) was found to be the most effective (entries 8–12). In addition, other silver additives were found to promote this coupling reaction (entries 13 and 14) and AgNTf2 gave the best yield of 62%. Atmosphere also has a great impact on this reaction (entries 15–17) and the yield was superior to the air and oxygen atmosphere under nitrogen atmosphere. When the ratio of reactants was 1:2 (1a: 2a), a yield of 76% was given. Considering that the divalent copper salt might act as an oxidant in the reaction, the co-additive was replaced with an equal amount of single copper acetate to obtain 3aa in 79% yield (entry 18). Reducing the loading of copper acetate to 2 equiv. resulted in 3aa in 82% yield (entries 19 and 20). Among other substituted acetophenones (entries 21 and 22), α-Br acetophenone led to a lower yield of 31% and α-OTs gained 3aa in 68% yield. Finally, the optimized reaction conditions were identified as follows: azobenzene 1a (0.20 mmol), α-Cl acetophenone 2a (0.40 mmol), [Cp*RhCl2]2 (5.0 mol%), AgNTf2 (20 mol%) and Cu(OAc)2 (2.0 equiv.) in DCE at 110 ℃ for 24 h under nitrogen.
|
|
Table 1 Optimization of the reaction conditions.a |

{kind=link}
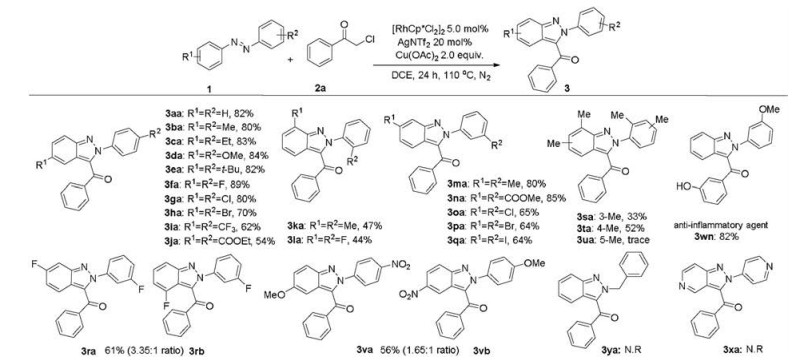
Under the optimized conditions (Table 1, entry 19), we first studied the scope and generality of various azobenzenes as shown in Scheme 1. Azobenzenes bearing various electron-donating/withdrawing groups and halides at the para position all coupled smoothly with 2a to afford the target products in generally good yields (3aa-3ja). Among them, the reaction of various alkyl or alkoxy or halogen-substituted azobenzenes with 2a gained up to 89% good yields. Electron-withdrawing group led to the moderate yields (3ia-3ja). The ortho-substituted azobenzenes gave the annulated products 3ka and 3la in 47% and 44% yields. In the case of meta-substituted substrates, the C-H activation occurred selectively and consistently at less hindered site, furnishing the products in 64%–85% yields (3ma-3qa). It is noteworthy that meta-fluoro substituent resulted in a mixture of two regioisomers (3ra and 3rb) in 61% combined yield with 3.3:1 ratio, favoring functionalization at the less hindered position. In addition, the disubstituted azobenzenes gave the target products in 33% and 52% yields (3sa-3ta). However, 3ua did not get the corresponding target product, perhaps due to the large steric hindrance. In this case of unsymmetrical azobenzenes, the ( E)-1-(4-methoxyphenyl)-2-(4-nitrophenyl)diazene led to the indazole products in 56% total yield with 1.65:1 regioselectivity. Simultaneously, 1-(3-methoxyphenyl)-2-phenyldiazene was coupled with 2-chloro-1-(3-hydroxy phenyl)ethan-1-one to obtain a single isomer 3wn with high regioselectivity and excellent yield (82%), which was used as an anti-inflammatory agent (Scheme 1) [2b]. Alkyl or heterocyclic azo compounds did not give the target product the under standard conditions (3xa-3ya).

|
Download:
|
| Scheme 1. Scope of azobenzenes. Reaction conditions: 1 (0.20 mmol), 2a (0.40 mmol), [Cp*RhCl2]2 (5.0 mol%), AgNTf2 (20 mol%), and Cu(OAc)2 (2.0 equiv.) in DCE (2.0 mL) at 110 ℃ for 24 h under nitrogen. Isolated yields. | |
{kind=link}
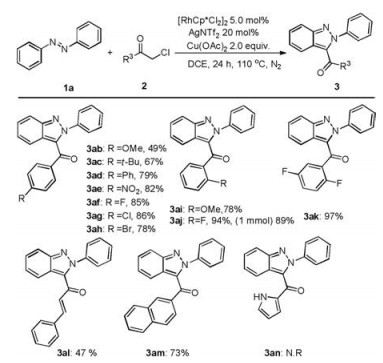
Meanwhile, the scope of α-Cl ketones was studied as shown in Scheme 2. First, diverse monosubstituted α-Cl ketones (2b-2j) and disubstituted α-Cl ketones (2k) were selected as the substrates for this reaction, furnishing the desired products 3ab-3ak in moderate to high yields. These results revealed that the electron density of the benzene ring had effect on the coupling reaction, in which strong electron-withdrawing groups (3ae and 3ak) appear to be beneficial for this transformation. The coupling reaction also exhibited better functional group tolerance (F, Cl, Br, Ph, NO2) in 78%–97% yields. Significantly, 3aj could be obtained in 89% yield when the reaction was enlarged as the scale of 1.0 mmol. Furthermore, alkenyl- and naphthyl-substituted analogues also worked well under the standard reaction conditions, leading to the corresponding 3-acyl-2H-indazoles in 47% (3al) and 73% (3am) yields. However, 2-chloro-1-(1 H-pyrrol-2-yl)ethan-1-one did not undergo the annulation (3an).

|
Download:
|
| Scheme 2. Scope of α-Cl ketones. Reaction conditions: 1a (0.20 mmol), 2 (0.40 mmol), [Cp*RhCl2]2 (5.0 mol%), AgNTf2 (20 mol%), and Cu(OAc)2 (2.0 equiv.) in DCE (2.0 mL) at 110 ℃ for 24 h under nitrogen, Isolated yields. | |
{kind=link}
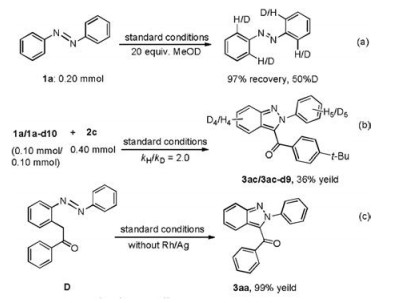
To clarify the reaction mechanism, some control experiments were conducted. When azobenzene 1a reacted with MeOD, azobenzene was recovered in 97% yield (Scheme 3a). 50% H/D exchange was found, which revealed that the C-H activation was reversible. A kinetic isotope ( kH/kD=2.0) was revealed in deuterium competition experiment (see Supporting information), implying that the C-H bond activation process was probably involved as the rate-determining step (Scheme 3b). Morever, α-aryl-ketones species D could be converted to 3aa in the absence of the catalyst (Scheme 3c). It revealed that D might be an key intermediate in the cyclization process (Supporting information). Furthermore, a radical trapping experiment suggested that radical process could be ruled out (Supporting information).

|
Download:
|
| Scheme 3. Mechanism studies. | |
{kind=link}
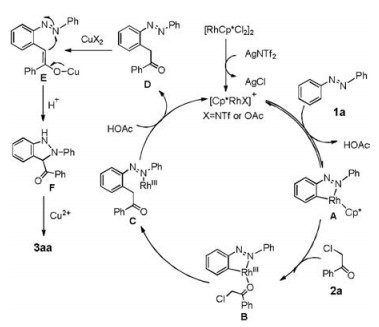
On the basis of the above experiments and literature reports [6a, 7], a plausible mechanism is proposed as shown in Scheme 4. Coordination of 1a with a cationic Rh(Ⅲ) species followed by C-H bond activation affords rhodacycle intermediate A. Subsequent coordination and migratory insertion of α-Cl acetophenone 2a produces intermediate C [9a], which undergoes ligand exchange to form α-aryl-ketones species D and a regenerated Rh(Ⅲ) species for next cycle. Next, Cu-mediated enol-tautomerization and intra molecular cyclization produced the intermediate E and F [6k], which was subsequently oxidized by Cu(Ⅱ) to provide 3-acyl-2H-indazole 3aa.

|
Download:
|
| Scheme 4. Proposed reaction mechanism. | |
{kind=link}
In summary, we have developed an efficient protocol to access 3-acyl-2H-indazoles via Rh(Ⅲ)-catalyzed C-H activation/annulation starting from readily available azobenzenes and α-Cl ketones. Various indazoles were conveniently afforded in good to excellent yields under the relatively mild conditions. It represents a novel and efficient method to access such a framework from simple starting materials.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21572072), 111 Project (No. BC 2018061), and Xiamen Southern Oceanographic Center (No. 15PYY052SF01).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.12.027.
| [1] |
(a) J.D. Rodgers, B.L. Johnson, C.W. Chang, et al., Bioorg. Med. Chem. Lett. 6 (1996) 2919-2924; (b) S. Bräse, C. Gil, K. Knepper, Bioorg. Med. Chem. Lett. 10 (2002) 2415-2437; (c) H. Cerecetto, A. Gerpe, M. Gonza'lez, V.J. Ara'n, C.O. de Oca'riz, Mini-Rev. Med. Chem. 5 (2005) 869-878; (d) J. Magano, M. Waldo, D. Greene, E. Nord, Org. Process Res. Dev. 12 (2008) 877-883; (e) M.J. Haddadin, W.E. Conrad, M.J. Kurth, Mini-Rev. Med. Chem. 12 (2012) 1293-1300. |
| [2] |
(a) L.J. Huang, M.L. Shih, H.S. Chen, et al., Bioorg. Med. Chem. 14 (2006) 528-536; (b) R.J. Steffan, E.M. Matelan. WO 2006/050006A2, 2006; (c) R. Halim, M. Harding, R. Hufton, et al., WO 2012/051659A1, 2012; (d) Y. Jia, J. Zhang, J. Feng, et al., Chem. Biol. Drug Des. 83 (2014) 306-316; (e) W. Aman, J. Lee, M. Kim, et al., Med. Chem. Lett. 26 (2016) 1188-1192 |
| [3] |
(a) M.D. Angelis, F. Stossi, K.A. Carlson, B.S. Katzenellenbogen, J.A. Katzenellenbogen, J. Med. Chem. 48 (2005) 1132-1144; (b) G. Luo, L. Chen, G. Dubowchik, J. Org. Chem. 71 (2006) 5392-5395; (c) A. Bunnell, C. O'Yang, A. Petrica, M.J. Soth, Synth. Commun. 36 (2006) 285-293; (d) C. Wu, Y. Fang, R.C. Larock, F. Shi, Org. Lett. 12 (2010) 2234-2237; (e) M.R. Kumar, A. Park, N. Park, S. Lee, Org. Lett. 13 (2011) 3542-2545; (f) A. Unsinn, P. Knochel, Chem. Commun. 48 (2012) 2680-2682; (g) W.S. Yong, S. Park, H. Yun, P.H. Lee, Adv. Synth. Catal. 358 (2016) 1958-1967; (h) Z.A. Henley, B.D. Bax, L.M. Inglesby, et al., ACS Med. Chem. Lett. 8 (2017) 1093-1098; (i) J.S. Zhu, N. Kraemer, M.E. Shatskikh, et al., Org. Lett. 20 (2018) 4736-4739; (j) Q. Gao, X. Han, P. Tong, et al., Org. Lett. 21 (2019) 6074-6078. |
| [4] |
(a) L. Ackermann, Chem. Rev. 111 (2011) 1315-1345; (b) S.H. Cho, J.Y. Kim, J. Kwak, S. Chang, Chem. Soc. Rev. 40 (2011) 5068-5083; (c) N. Kuhl, M.N. Hopkinson, J. Wencel-Delord, F. Glorius, Angew. Chem. Int. Ed. 51 (2012) 10236-10254; (d) G. Rouquet, N. Chatani, Angew. Chem. Int. Ed. 52 (2013) 11726-11743; (e) S.D. DeSarkar, W. Liu, S.I. Kozhushkov, L. Ackermann, Adv. Synth. Catal. 356 (2014) 1461-1479; (f) S.A. Girard, T. Knauber, C.J. Li, Angew. Chem. Int. Ed. 53 (2014) 74-100; (g) O. Daugulis, J. Roane, L.D. Tran, Acc. Chem. Res. 48 (2015) 1053-1064; (h) Z. Huang, H.N. Lim, F. Mo, M.C. Young, G. Dong, Chem. Soc. Rev. 44 (2015) 7764-7786; (i) Z. Chen, B. Wang, Y. Zhang, et al., Org. Chem. Front. 2 (2015) 1107-1295; (j) W. Liu, L. Ackermann, ACS Catal. 6 (2016) 3743-3752; (k) C. Sambiagio, D. Schonbauer, M. Schnürch, et al., Chem. Soc. Rev. 47 (2018) 6603-6743; (l) X. Han, P.P. Lin, Q. Li, Chin. Chem. Lett. 30 (2019) 1495-1502. |
| [5] |
(a) T. Zhou, B. Li, B. Wang, Org. Lett. 22 (2016) 5066-5069; (b) P. Hu, Y. Zhang, B. Liu, X. Li, Org. Chem. Front. 5 (2018) 3263-3266; (c) L. Wang, D. Xiong, L. Jie, C. Yu, X. Cui, Chin. Chem. Lett. 29 (2018) 907-910; (d) Z. Shen, C. Pi, X. Cui, Y. Wu, Chin. Chem. Lett. 30 (2019) 1374-1378; (e) X. Song, B.N.D. Doan, X. Zhang, R. Lee, X. Fan, Org. Lett. 2 (2020) 46-51. |
| [6] |
(a) Y. Lian, R.G. Bergman, L.D. Lavis, J.A. Ellman, J. Am. Chem. Soc. 135 (2013) 7122-7125; (b) D. Zhao, Q. Wu, X. Huang, et al., Chem. Eur. J. 19 (2013) 6239-9244; (c) C. Qian, D. Lin, Y. Deng, et al., Org. Biomol. Chem. 12 (2014) 5866-5875; (d) J.R. Hummel, J.A. Ellman, J. Am. Chem. Soc. 137 (2015) 490-498; (e) S. Sharma, S.H. Han, I.S. Kim, et al., Org. Lett. 17 (2015) 2852-2855; (f) X. Geng, C. Wang, Org. Lett. 17 (2015) 2434-2467; (g) T. Jeong, S.H. Han, et al., Org. Lett. 18 (2016) 232-235; (h) G. Hong, A.N. Aruma, X. Zhu, S. Wu, L. Wang, Synthesis 48 (2016) 1147-1158; (i) Z. Long, Y. Yang, J. You, Org. Lett. 19 (2017) 2781-2784; (j) S. Cai, S. Lin, X. Yi, C. Xi, J. Org. Chem. 82 (2017) 512-520; (k) Z. Long, Z. Wang, D. Zhou, D. Wan, J. You, Org. Lett. 19 (2017) 2777-2780; (l) X. Chen, G. Zheng, G. Song, X. Li, Adv. Synth. Catal. 360 (2018) 2836-2842; (m) R. Chun, S. Kim, I.S. Kim, et al., Adv. Synth. Catal. 361 (2019) 1-11; (n) J. Zhu, S. Sun, J. Cheng, Tetrahedron Lett. 59 (2018) 2284-2287; (o) H. Oh, S. Han, A.K. Pandey, et al., J. Org. Chem. 83 (2018) 4070-4077. |
| [7] |
D.G. Yu, F. de Azambuja, F. Glorius, Angew. Chem. Int. Ed. 53 (2014) 2754-2758. DOI:10.1002/anie.201310272 |
| [8] |
J. Li, Z. Zhang, M. Tang, X. Zhang, J. Jin, Org. Lett. 18 (2016) 3898-3901. DOI:10.1021/acs.orglett.6b01916 |
| [9] |
(a) C. Yang, F. Song, J. Chen, Y. Huang, Adv. Synth. Catal. 359 (2017) 3496-3502; (b) C. Xie, Z. Dai, Y. Niu, C. Ma, J. Org. Chem. 83 (2018) 2317-2323; (c) R.A. Bohmann, J.H. Schoebel, Y. Unoh, M. Miura, C. Bolm, Adv. Synth. Catal. 361 (2019) 2000-2003; (d) H. Liu, C. Wu, H. Liu, J. Wang, J. Org. Chem. 84 (2019) 13262-13275. |
| [10] |
J. Zhou, J. Li, H. Liu, et al., Org. Lett. 20 (2018) 7645-7649. DOI:10.1021/acs.orglett.8b03383 |
| [11] |
(a) S. Patai, Z. Rappoport, The Chemistry of α-Haloketones, α-Haloaldehydes and α-Haloimines, Wiley, New York, 1988, pp. 1-489; (b) W.A. Erian, M.S. Sherif, M.H. Gaber, Molecules 8 (2003) 793-865. |
| [12] |
(a) L. Xu, L. Wang, X. Cui, et al., Org. Lett. 19 (2017) 4343-4346; (b) Y. Feng, N. Tian, Y. Li, et al., Org. Lett. 19 (2017) 1658-1661; (c) Y. Yu, Y. Feng, R. Chauvin, et al., Org. Lett. 20 (2018) 4209-4212; (d) Z. Yang, Z. Song, L. Wang, L. Jie, X. Cui, Chem. Commun. 55 (2019) 6094-6097; (e) B. Wu, Z. Yang, H. Zhang, L. Wang, X. Cui, Chem. Commun. 55 (2019) 4190-4193; (f) S. Du, C. Pi, T. Wan, Y. Wu, X. Cui, Adv. Synth. Catal. 361 (2019) 1766-1770; (g) S. Huang, H. Li, X. Sun, et al., Org. Lett. 21 (2019) 5570-5774; (h) J. Ren, X. Yan, X. Cui, et al., Green Chem. 22 (2020) 265-269; (i) Y. Wu, C. Pi, X. Cui, Y. Wu, Org. Lett. 22 (2020) 361-364; (j) Y. He, C. Pi, Y. Wu, X. Cui, Chin. Chem. Lett. 31 (2020) 396-400. |