 2021, Vol. 32
2021, Vol. 32 


b Key Laboratory of Optic-electric Sensing and Analytical Chemistry for Life Science, MOE, State Key Laboratory Base of Eco-Chemical Engineering, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
Organosulfur compounds have drawn considerable attentions, because of their remarkably bioactivities in medicinal chemistry [1], as well as their widespread applications in material science [2]. Therefore, the construction of C–S bonds is a fundamental process in the synthesis of sulfur-containing molecules [3]. Transition metal-catalyzed cross-coupling reactions between thiols, arylboronic acids or pseudo-halides with disulfides or organic halides have been broadly used as a general method for the construction of C–S bonds [4]. On the other hand, C–H bonds functionalization has been widely regarded as an alternative approach to traditional cross-coupling reactions due to its high efficiency and atom economy [5]. In recent years, tremendous progress has been made for the construction of C–S bonds through C–H bond functionalization [6]. However, these methods inevitably involve using odorous sulfur sources and usually harsh reaction conditions were required. Elemental sulfur exists in the form of sulfur powder which was considered as non-poisonous and tractable reagent compared to thiols [7]. Over the last several years, elemental sulfur has emerged as an ideal sulfur surrogate in the organic transformations [8]. However, challenges still remain, and it is highly desirable to develop more efficient and convenient strategies for the synthesis of sulfur-containing compounds using sulfur powder as sulfur source.
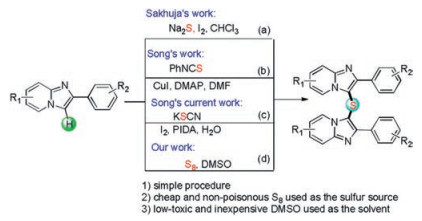
Imidazo[1, 2-a]pyridines are readily accessible heterocycles that have found widely applications across organic chemistry, materials science, medicinal chemistry, and chemical biology [9]. Compounds with an imidazo[1, 2-a]pyridine motifs are well known to possess various pharmacological activities. Most importantly, some commercially available drugs contain this core structure, such as minodronic acid (used to treat osteoporosis) [10], zolmidine (gastroprotective agent) [11] and zolpidem (used to treat insomnia) [12]. Importantly, some C-3 sulfenylation imidazo-[1, 2-a]pyridines possess medical activities, such as the inhibitor of human rhinovirus I [13], antitumor agent II [14], and antiosteoporotic agent III [15] (Fig. 1). As a consequence, in the past few decades, significant efforts have been made toward to the synthesis and modification of imidazo[1, 2-a]pyridines [16]. Recently, many methods have been developed for C-3 functionalization of imidazo[1, 2-a]pyridines [17]. On the other hand, biheteroaryl skeletons have been widely known as prominent heterocycles possessing various biological, and pharmacological activities [18]. In consideration of the biological activities of biheteroaryl scaffolds and imidazopyridine derivatives in medicinal chemistry, extensive efforts have been devoted to the development of efficient methods for their synthesis and functionalization. In 2017, Sakhuja et al. disclosed that bis-(imidazo[1, 2-a]pyridin-3-yl)sulfanes could be obtained in high yields when treatment of imidazo[1, 2-a]pyridines with Na2S by using iodine as a green catalyst (Scheme 1a) [17f]. In 2019, Songet al. reported an elegant Cu(I)-catalyzed double thiolation of imidazopyridines for the construction of bis(imidazo[1, 2-a]pyridin-3-yl)sulfanes using isothiocyanate as the sulfur source (Scheme 1b) [17e]. Although these reactions have made some great advantages, the development of a convenient and green method that can complement existing synthetic methods remains an ongoing challenge. To the best of our knowledge, double C–S bonds forming reactions at C-3 position of imidazo[1, 2-a]pyridines using environmentally benign sulfur powder as sulfur surrogate under metal-free condition is rarely reported. With our growing interest in the synthesis of sulfur-containing compounds [19], herein, we report an efficient, practical and simple method for the synthesis of bis(imidazo[1, 2-a]pyridin-3-yl)sulfanes using S8 as sulfur source under metal-free conditions (Scheme 1d). Notably, during the submission process of our manuscript, Song and co-workers developed a novel approach to bis(imidazo[1, 2-a]-pyridin-3-yl)sulfanes through iodine catalyzed chalcogenation of imidazopyridines and KSCN (Scheme 1c) [20].

|
Download:
|
| Fig. 1. Bioactive molecules containing imidazo[1, 2-a]pyridine frameworks. | |

|
Download:
|
| Scheme 1. Recent strategies for the synthesis of bis(imidazo[1, 2-a]pyridin-3-yl)sulfanes. | |
Initially, the reaction was carried out by treatment of imidazo[1, 2-a]pyridines 1a with sulfur powder in 2 mL DMF at 120 ℃ under air atmosphere (Table 1). To our delight, the desired product 2a was isolated in 67% yield (entry 1). To enhance the reaction efficiency, other solvents were then examined. As shown in Table 1, three solvents including DMSO, toluene, THF (entries 2–4) were investigated, and DMSO was found to be the most effective solvent, giving desired product 2a in 87% yield (entry 2). We also tested various sulfur sources, and sulfur powder showed the best result (compare with entries 2, 5–7). A series control experiments indicated that the reaction did not take place when Na2S or Na2S·9H2O were used as the sulfur source. However, when K2S was used in the present transformation, the reaction could proceed and delivered a 53% yield. Interestingly, the yield decreased to 61% under N2 atomsphere (entry 8), which showed that O2 plays an important role in the present transformation. Finally, decreasing the reaction temperature resulted in a lower conversion (entries 9 and 10).
|
|
Table 1 Optimization of reaction conditions.a |

{kind=link}
{kind=link}
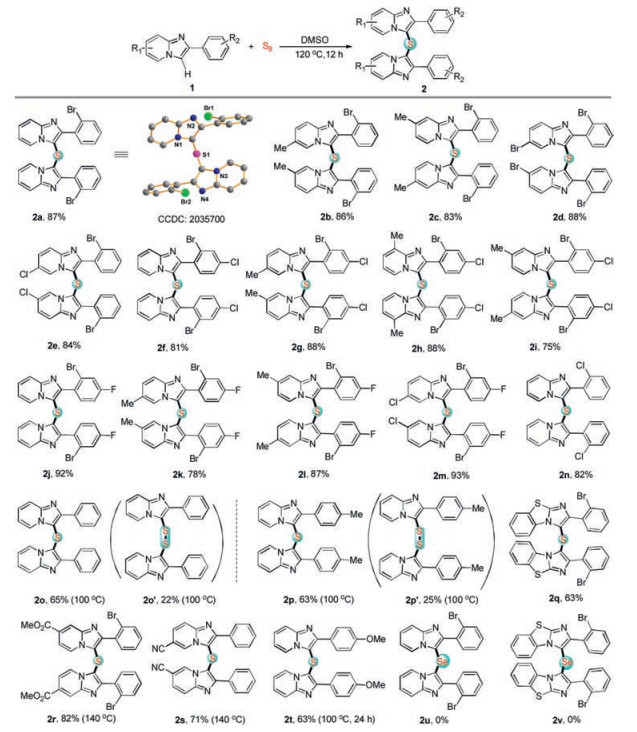
With the optimal conditions in hand, we next proceeded to explore the substrate scope with the corresponding imidazo[1, 2-a]-pyridines (Scheme 2). To our delight, most of the substrates provided the corresponding products in moderate to good yields. The structures of the products were unambiguously confirmed by the single crystal X-ray diffraction of compound (2a). Both electron-withdrawing and electron-donating groups were found to show no significant difference in reactivity in the present transformation. Various functional groups such as fluorine, chlorine, bromo and methyl group were well tolerated with the optimum conditions, providing more chances for further functionalization or modification. In addition, −COOMe, -CN and -OMe were compatible in present transformation. Notably, 2-(2-bromophenyl)benzo[d]imidazo[2, 1-b]thiazole also afforded the desired product 2q in 63% yield. Analyzing substrates from the perspective of steric hindrance effect, 2-phenylimidazo[1, 2-a]pyridine derivatives bearing Br atom at C-2 position on the benzene ring should have lower reactivity in the transformation. Unexpectedly, these derivatives showed higher reactivity than other types of imidazo-[1, 2-a]pyridines. It is interesting to note that disulfide products 2o' and 2p' were obtained when 1o and 1p were tested with sulfur powder at 100 ℃.

|
Download:
|
| Scheme 2. Reaction scope of imidazo[1, 2-a]pyridines with sulfur powder. Reaction conditions: imidazo[1, 2-a]pyridine 1 (0.2 mmol), sulfur powder (0.4 mmol), DMSO (2 mL), under air atmosphere, at 120 ℃ for 12 h. Isolated yield. | |
{kind=link}
In addition, the feasibility of the synthesis method in gram level was also investigated (Scheme 3). To our delight, the present reaction could afford 1.29 g of 2a under the standard conditions, with no significant loss of its efficiency.

|
Download:
|
| Scheme 3. Synthesis of 2a on a gram scale. | |
{kind=link}
To acquire insights into this oxidative dual C–H sulfenylation reaction, control experiment was carried out (Scheme 4). The reaction proceed smoothly in the presence of a radical scavenger TEMPO, which preliminarily indicates that this transformation might not involve a radical process (Scheme 4a). When 1o reacted with 1o' under the standard conditions, the desired product 2o was obtained in 95% yield, which suggests that disulfides might be a key intermediate in this transformation (Scheme 4b).

|
Download:
|
| Scheme 4. Control experiments. | |
{kind=link}
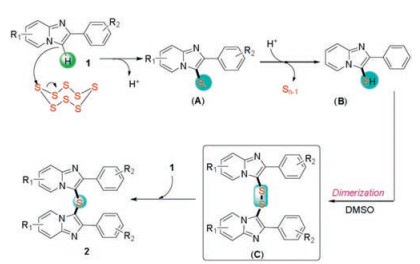
On the basis of the preliminary results and the previous literature reports [21], a plausible mechanism was proposed inScheme 5. The initial transformation would be the activation of elemental sulfur by nucleophilic attack of 1 on the S8 ring to generate intermediate (A), which could be changed into 2-phenylimidazo[1, 2-a]pyridine-3-thiol (B). Subsequently, the intermediate (B) was oxidized to disulfide (C) by using DMSO as the oxidant. Finally, the imidazo[1, 2-a]pyridines attacked the disufides (C) resulting the desired product 2.

|
Download:
|
| Scheme 5. Proposed mechanism for the direct transformation. | |
{kind=link}
In summary, we have successfully developed a novel, practical and easy-handle method for the synthesis of heteroaromatic sulfides with potentially biological activities using sulfur powder as a sulfur source. Notably, the developed method could be carried out using cheap and abundantly chemicals, without the need of a transition metal catalyst, as well as strong oxidants or bases, thus facilitating benchtop reactions that could be performed in a simple fashion. Therefore, the protocol will attract much attention in organic chemistry and medicinal chemistry. Further investigation of the bioactivities of these compounds is ongoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financialinterestsor personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21302110), the Natural Science Foundation of Shandong Province (No. ZR2016JL012), and the Scientific Research Foundation of Qingdao University of Science and Technology.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.12.046.
| [1] |
(a) M.D. McReynolds, J.M. Dougherty, P.R. Hanson, Chem. Rev. 104 (2004) 2239-2258; (b) H. Liu, X.F. Jiang, Chem. Asian J. 8 (2013) 2546-2563. |
| [2] |
C. Shen, P.F. Zhang, Q. Sun, et al., Chem. Soc. Rev. 44 (2015) 291-314. DOI:10.1039/C4CS00239C |
| [3] |
(a) A. Ghaderi, Tetrahedron 72 (2016) 4758-4782; (b) X.M. Xu, D.M. Chen, Z.L. Wang, Chin. Chem. Lett. 31 (2020) 49-57; (c) L.L. Wang, M. Zhang, Y.L. Zhang, et al., Chin. Chem. Lett. 31 (2020) 67-70; (d) Y.Q. Jiang, J. Li, Z.W. Feng, et al., Adv. Synth. Catal. 362 (2020) 2609-2614; (e) Z. Wang, W.M. He, Chin. J. Org. Chem. 39 (2019) 3594-3595. |
| [4] |
I.P. Beletskaya, V.P. Ananikov, Chem. Rev. 111 (2011) 1596-1636. DOI:10.1021/cr100347k |
| [5] |
(a) Z. Gan, Q. Yan, G. Li, et al., Adv. Synth. Catal. 361 (2019) 4558-4567; (b) X. Wang, Q. Wang, Y. Xue, et al., Chem. Commun. 56 (2020) 4436-4439. |
| [6] |
C. Ravi, S. Adimurthy, Chem. Rec. 17 (2017) 1019-1038. DOI:10.1002/tcr.201600146 |
| [7] |
(a) F.J. Chen, G. Liao, X. Li, J. Wu, B.F. Shi, Org. Lett. 16 (2014) 5644-5647; (b) C. Chen, L.L. Chu, F.L. Qing, J. Am. Chem. Soc. 134 (2012) 12454-12457. |
| [8] |
T.B. Nguyen, Adv. Synth. Catal. 359 (2017) 1066-1130. DOI:10.1002/adsc.201601329 |
| [9] |
(a) L. Dyminska, Bioorg. Med. Chem. 23 (2015) 6087-6099; (b) A.J. Stasyuk, M. Banasiewicz, M.K. Cyranski, D.T. Gryko, J. Org. Chem. 77 (2012) 5552-5558; (c) H. Iida, R. Demizu, R. Ohkado, J. Org. Chem. 83 (2018) 12291-12296; (d) S. Ulloora, A.V. Adhikari, R. Shabaraya, Chin. Chem. Lett. 24 (2013) 853-856. |
| [10] |
L.A. Sorbera, J. Castaner, P.A. Leeson, Drugs Future 27 (2002) 935-941. DOI:10.1358/dof.2002.027.10.701186 |
| [11] |
L. Almirante, L. Polo, A. Mugnaini, et al., J. Med. Chem. 8 (1965) 305-312. DOI:10.1021/jm00327a007 |
| [12] |
S.Z. Langer, S. Arbilla, J. Benavides, B. Scatton, Adv. Biochem. Psychopharmacol. 46 (1990) 61. |
| [13] |
C. Hamdouchi, J. de Blas, M. del Prado, et al., J. Med. Chem. 42 (1999) 50-59. DOI:10.1021/jm9810405 |
| [14] |
T. Guo, X.N. Wei, M. Zhang, et al., Chem. Commun. 56 (2020) 5751-5754. DOI:10.1039/D0CC00043D |
| [15] |
Y. Kawai, S. Satoh, H. Yamazaki, N. Kayakiri, K. Yoshihara, T. Oku, Patent, WO-9634866-A1, 1995.
|
| [16] |
(a) J. Koubachi, S.E. Kazzouli, M. Bousmina, G. Guillaumet, Eur. J. Org. Chem. 24 (2014) 5119-5138; (b) Q. Cai, M.C. Liu, B.M. Mao, et al., Chin. Chem. Lett. 26 (2015) 881-884; (c) A.K. Bagdi, S. Santra, K. Monir, A. Hajra, Chem. Commun. 51 (2015) 1555-1575; (d) A.K. Bagdi, M. Rahman, S. Santra, A. Majee, A. Hajra, Adv. Synth. Catal. 355 (2013) 1741-1747; (e) X.Y. Li, Y. Liu, X.L. Chen, et al., Green Chem. 22 (2020) 4445-4449; (f) S. Santra, A.K. Bagdi, A. Majee, A. Hajra, Adv. Synth. Catal. 355 (2013) 1065-1070; (g) K. Groebke, L. Weber, F. Mehlin, Synlett 6 (1998) 661-663; (h) C. He, J. Hao, H. Xu, et al., Chem. Commun. 48 (2012) 11073-11075; (i) F. Gao, K. Sun, X.L. Chen, et al., J. Org. Chem. 85 (2020) 14744-14752; (j) G. Kibriya, A.K. Bagdi, A. Hajra, J. Org. Chem. 83 (2018) 10619-10626; (k) M. Singsardar, S. Mondal, S. Laru, A. Hajra, Org. Lett. 21 (2019) 5606-5610; (l) L.J. Ma, X.P. Wang, W. Yu, B. Han, Chem. Commun. 47 (2011) 11333-11335. |
| [17] |
(a) H. Cao, S. Lei, N.Y. Li, et al., Chem. Commun. 51 (2015) 1823-1825; (b) G. Kibriya, S. Samanta, S. Jana, S. Mondal, A. Hajra, J. Org. Chem. 82 (2017) 13722-13727; (c) Y.J. Guo, S. Lu, L.L. Tian, et al., J. Org. Chem. 83 (2018) 338-349; (d) F. Shibahara, T. Kanai, E. Yamaguchi, et al., Chem. Asian J. 9 (2014) 237-244; (e) L.L. Tian, S. Lu, Z.H. Zhang, et al., J. Org. Chem. 84 (2019) 5213-5221; (f) S.M. Abdul Shakoor, D.S. Agarwal, S. Khullar, S.K. Mandal, R. Sakhuja, Chem. Asian J. 12 (2017) 3061-3068; (g) M. Yadav, S. Dara, V. Saikam, et al., Eur. J. Org. Chem. 29 (2015) 6526-6533; (h) S. Mondal, S. Samanta, S. Jana, A. Hajra, J. Org. Chem. 82 (2017) 4504-4510; (i) G.H. Xiao, H. Min, Z.L. Zheng, G.B. Deng, Y. Liang, Chin. Chem. Lett. 29 (2018) 1363-1366; (j) K. Sun, X. Wang, C. Li, H. Wang, L. Li, Org. Chem. Front. 7 (2020) 3100-3119. |
| [18] |
M. Shiri, M.A. Zolfigol, H.G. Kruger, Z. Tanbakouchian, Chem. Rev. 110 (2000) 2250-2293. DOI:10.1016/B978-0-12-385464-3.00002-9 |
| [19] |
(a) G. Li, Z. Gan, K. Kong, X. Dou, D. Yang, Adv. Synth. Catal. 361 (2019) 1808-1814; (b) G. Li, Q. Yan, X. Gong, X. Dou, D. Yang, ACS Sustainable Chem. Eng. 7 (2019) 14009-14015; (c) G. Li, G. Zhang, X. Deng, et al., Org. Biomol. Chem. 16 (2018) 8015-8019; (d) X. Gong, G. Li, Z. Gan, et al., Asian J. Org. Chem. 8 (2019) 1472-1478; (e) G. Li, Q. Yan, Z. Gan, Q. Li, D. Yang, Org. Lett. 21 (2019) 7938-7942; (f) Z. Gan, G. Li, X. Yang, et al., Sci. China Chem. 63 (2020) 1652-1658. |
| [20] |
Y.S. Zhu, Y.T. Xue, W.N. Liu, et al., J. Org. Chem. 85 (2020) 9106-9116. DOI:10.1021/acs.joc.0c01035 |
| [21] |
(a) T.B. Nguyen, L. Ermolenko, P. Retailleau, A. Al-Mourabit, Angew. Chem. Int. Ed. 53 (2014) 13808-13812; (b) H. Xie, J. Cai, Z. Wang, H. Huang, G.J. Deng, Org. Lett. 18 (2016) 2196-2199; (c) Y. Yang, W. Li, B. Ying, et al., ChemCatChem 8 (2016) 2916-2919. |