 2021, Vol. 32
2021, Vol. 32 


b International College, Zhengzhou University, Zhengzhou 450052, China
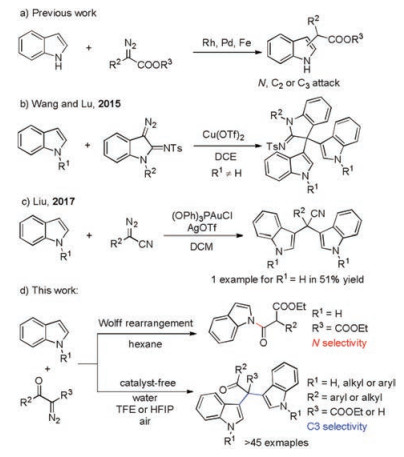
Bis(indolyl)methane (BIM) is a significant structural motif isolated from diverse natural sources [1] and exhibits broad spectrum bioactivities [2]. Traditional construction of such a skeleton bases on Friedel-Crafts reaction of indoles with carbonyl compounds (aldehydes and ketones) in the presence of Lewis acid as a catalyst [2f]. To avoid the catalyst and additives, greener procedures have been explored recently [3]. However, most products obtained are BIMs with tertiary carbon. Molecules containing quaternary carbon center are usually problematic to be prepared, but show unique bioactivities due to its special rigid three-dimensional structure [4]. Thus, a greener procedure to access BIMs with quaternary carbon is highly desired.
On the other hand, the condensation of indoles and diazo compounds via transition metal catalyzed carbenoid insertion to construct C-C bond has been extensively studied. Generally, all of the N, C2 and C3 atoms could work as nucleophilic site for N-H free indoles (Scheme 1a). Selective C3 attack usually was achieved with N- and C2- substituted indoles [5]. Very recently, Wang, Lu group [6] reported a copper catalyzed C3-bisindolation of 3-diazoindolin-2-imines with N substituted indoles (Scheme 1b). However, N-H free indoles were not effective. Soon after, Liu group [7] developed a gold catalyzed bisindolylation of aryldizao cyanide and only one example of N-H free indole was used as substrate with moderate yield (Scheme 1c). Herein, we disclose a novel reaction of N-H free indoles with α-diazo-β-ketoesters under metal and additives free conditions. This procedure provides a greener protocol to afford various BIMs with quaternary carbon (Scheme 1d). Molecular oxygen from air as an oxidant, water and fluorinated alcohol as co-solvent and promoter make this procedure greener. Catalyst and additives were avoided and only N2 and water were produced as by-products. This high atom economy reaction benefits sustainable development.

|
Download:
|
| Scheme 1. Divergent reactions of indole and diazo compounds. | |
We initiated our study using indole (1a) and ethyl 2-diazo-3-oxobutanoate (2a) as the model substrates to screen various reaction parameters (Table 1). When non-polar solvents were used, Wolff rearrangement [8] reaction occurred, affording 4a as a main product (entries 1–4). The product 4a was obtained in 82% yield in hexane at 120 ℃ under air atmosphere for 12 h (entry 4). No reaction occurred in EtOH or MeCN (entries 5 and 6). Water is an ideal solvent in organic synthesis attribute to its being green, cheap, nontoxic and non-flammable [9], moreover could accelerate reaction rate owing to its hydrophobic effect [10]. To our delight, water as solvent led to the product 3aa in 28% yield (entry 7). Then we tried to add other polar solvent in water to improve the solubility of substrates. EtOH or MeCN as co-solvent did not give better results (entries 8 and 9). Fluorinated alcohol, 1, 1, 1, 3, 3, 3-hexafluoroisopropanol (HFIP) and 1, 1, 1-trifluoroethanol (TFE) possess unique properties, such as high hydrogen bonding donor ability, low nucleophilicity, high ionizing power, extreme polarity, enhanced acidity, ability to stabilize cationic species and solvate water [11]. This kind of alcohol has been employed to promote diverse reactions including oxidation [11a-c] and Friedel-Crafts reactions [3a, 12]. Hence, we hypothesized that fluorinated alcohol might facilitate the desired reaction. As expected, 3aa was obtained in elevated yield (39%) when water and TFE were used as co-solvent in 1:1 ratio of volume (entry 10). None of 3aa was afforded under nitrogen atmosphere (entry 11). Oxygen atmosphere gave the similar result to air (entry 12). These results suggested that oxygen from air might play the key role in the reaction. Increasing the solvent to 3 mL, 79% isolated yield of 3aa was obtained (entry 13). 75% Yield of 3aa was attained when water and HFIP were employed as co-solvents (entry 15). Not any transformation was detected when TFE or HFIP as sole solvent (entry 16). The results obtained above indicated that water and fluorinated alcohol played synergic role to accelerate the reaction.
|
|
Table 1 Optimization of the reaction conditions.a |

Under the optimal conditions in hand, scope of indoles was surveyed (Scheme 2, detailed experimental procedure see Supporting information). In general, electron-rich indoles worked well when either TFE or HFIP as co-solvent with water (3ba-3ga). In particularly, methoxy at different position of indoles all gave the corresponding products in good to excellent yields (3da-3fa, 81%–92%). For the indoles with halogen and electron-withdrawing groups, reactions proceeded smoothly in HFIP than TFE as co-solvent with water (3ha-3pa). F, Cl, Br and I were tolerated well and the desired products were obtained in good yields (3ha-3ma, 68%–86%). Ester or carboxyl group in the indole afforded the desired products in moderate yields (3na-3pa, 43%–55%) as well as tertiary alcohols (5na-5pa) as by-products. While, tertiary alcohols were given as the main products (5qa and 5ra) for indoles with strong electron-withdrawing groups (nitrile and nitro group). Subsequently, N-substituted indoles were investigated. Both N-alkyl and phenyl indoles led to bisindolylated products in good yields (3sa-3wa and 3ya, 67%–89%). While, N-tosyl indole did not work (3xa). In addition, 3aa was obtained in 78% for N-Boc indole. Fused indole also could provide the desired product in good yield (3ya).

|
Download:
|
| Scheme 2. Scope of indole derivatives. Reaction conditions: 1 (0.1 mmol), 2a (0.1 mmol) and solvent A or B at 120 ℃ under air atmosphere for 12 h. Isolated yields. Solvent A: H2O (2 mL) and TFE (1 mL); B: H2O (1 mL) and HFIP (0.5 mL). | |
Then scope of diazo compounds 2 was also investigated (Table 2). Apart from acetyl, α-diazo alkyl ketoesters with cyclopropyl, propyl and isopropyl also led to the corresponding products in moderate to good yields (3sb-3sd). Various para-and meta-substituted benzoyl diazos gave the products in good yields (3sf-3sk, 60%–82%). Yield of product 3sl (49%) was generated for α-diazo ortho-F aryl ketoesters. α-Diazo 2-furyl and 2-thienyl ketoesters provided the corresponding products in 70% (3sm) and 77% (3sn) yields, respectively. Monosubstituted α-diazo aryl ketone (2o) and ethyl diazoacetate (2p) produced 3so and 3sp with 1-methylindole in moderate yields. While, α-diazo-β-diketo (2r and 2u), α-diazo-β-diester (2s) and ethyl 2-diazo-2-phenylacetate (2t) failed to give the desired products, perhaps due to the reason that the O-H insertion intermediates could not be transformed to vicinal tricarbonyl compounds under air.
|
|
Table 2 Scope of diazo compounds.a |

This protocol was also applied to the synthesis of unsymmetric BIMs. The results are depicted in Scheme 3. In view of the fact that electron-rich indoles react faster than deficient ones, the loading of the comparative electron-deficient indole (1a, 1k or 1e) is slightly excessive than 1-methylindole (1s). As a result, unsymmetric BIMs 3saa, 3ska, 3sea and 3vea were produced in moderate yields (41%–57%). By utilizing the discrepant nucleophilicity of indoles with different substituents, 3zsa, 3zaa and 3zka were obtained in moderate to good yields (47%–68%). And 3eaa was provided in a 31% yield from similar nucleophilicity of 1e and 1a.

|
Download:
|
| Scheme 3. Synthesis of unsymmetric bis(indolyl)methanes. Reaction conditions: 1 (electron-rich indole, 0.1 mmol), 1 (electron-deficient indole, 0.15 mmol), 2a (0.2 mmol), H2O (2 mL) and TFE (1 mL) at 120 ℃ under air atmosphere for 12 h. Isolated yields. | |
To clarify the reaction mechanism, a series of control reactions were conducted. First, the reactions of 1-methylindole 1s and 6e were carried out in different solvents (Table 3). Mixed solvent of water and TFE led to the quantitative bisindolylated product 3se (entry 1). 6e might be a key intermediate from diazo compound 2e. Water or TFE as sole solvent resulted in lower yield of 3se (entries 2 and 3). These results demonstrate that combination of water and fluorinated alcohol paly the key role to accelerate reaction. Second, treatment of α-diazo-β-ketoester 2f in the mixed solvent of water and TFE under air produced the product 6f in 16% as well as Wolff rearrangement/decarboxylation product 7f in 69% yield (Scheme 4, entry 1). However, 6f was not detected in the water or TFE as sole solvent and only Wolff rearrangement products (7f or 8) were isolated (entries 2 and 3).
|
|
Table 3 Reaction of 1-methylindole and 6e in different solvents.a |

{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Scheme 4. Reaction of α-diazo-β-ketoesters in different solvents. Reaction conditions: 2f (0.1 mmol) and solvent at 120 ℃ under air atmosphere for 12 h. Isolated yields. | |
{kind=link}

|
(1) |
In addition, the reaction of 1s and ethyl 2-hydroxy-3-oxo-3-phenylpropanoate 9e was conducted under the standard reaction conditions. The desired product 3se was obtained in 89% yield (Eq. 1). The above results obtained imply that the diazo was oxidized by oxygen in the air, which was promoted by water and fluorinated alcohol. Generally, stoichiometric oxidant, such as tert-butyl hypochlorite [13], dimethyl dioxirane [14] or 2, 6-dichloropyridine N-oxides [15], is required for the oxidation of α-diazo-β-ketoester.
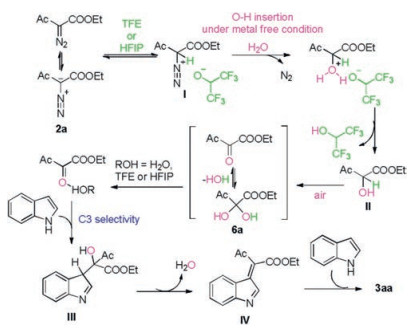
Based on the results above, a plausible reaction mechanism was proposed and depicted in Scheme 5. α-Diazo-β-ketoester 2a was protonated by fluorinated alcohol as Brønsted acid to afford the diazonium I [16]. Then nucleophilic substituent of diazonium I with water produced α-hydroxy-β-ketoester II as well as N2 [16, 17]. Subsequently, II was oxidized into 6a by oxygen molecule from air atmosphere [18]. Through this umpolung transformation, the nucleophile 2a was transformed as an electrophile 6a [19]. Subsequently, the in situ generated carbonyl group was activated by water or fluorinated alcohol with H-bond. Following by the nucleophilic addition of this carbonyl group with the indole gave III [20]. Intermediate IV was generated through elimination of one molecular H2O. Then the second nucleophilic attack of indole produced 3aa.

|
Download:
|
| Scheme 5. Plausible mechanism. | |
{kind=link}
In conclusion, we have developed a green, efficient and novel protocol to access BIMs with quaternary carbon starting from indoles and α-diazo-β-ketoesters. This approach is promoted by water and fluorinated alcohol as co-solvent under metal catalyst and additive free conditions. Only N2 and water were released as by-products. This high atom economy reaction benefits environmental protection and sustainable development. Moreover, a tandem reaction of O-H insertion, aerobic oxidation and bisindolylation were involved in this transformation and also applied in the synthesis of unsymmetric BIMs.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe greatly acknowledge financial support from the Ministry of Science and Technology of China (No. 2016YFE0132600), Henan Center for Outstanding Overseas Scientists (No. GZS2020001), National Innovation and Entrepreneurship Training Program for College students (No. 201910459064) and Zhengzhou University.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.12.026.
| [1] |
(a) T.R. Garbe, M. Kobayashi, N. Shimizu, et al., J. Nat. Prod. 63 (2000) 596-598; (b) M. Kobayashi, S. Aoki, K. Gato, et al., Chem. Pharm. Bull. 42 (1994) 2449-2451; (c) R. Veluri, I. Oka, I. Wagner-Döbler, H. Laatsch, J. Nat. Prod. 66 (2003) 1520-1523; (d) R. Bell, S. Carmeli, N. Sar, J. Nat. Prod. 57 (1994) 1587-1590. |
| [2] |
(a) C. Hong, G.L. Firestone, L.F. Bjeldanes, Biochem. Pharmacol. 63 (2002) 1085-1097; (b) T. Irie, K. Kubushiro, K. Suzuki, et al., Anticancer Res. 19 (1999) 3061-3066; (c) H.T. Le, C.M. Schaldach, G.L. Firestone, L.F. Bjeldanes, J. Biol. Chem. 278 (2003) 21136-21145; (d) A. Kamal, Y.V.V. Srikanth, M.N.A. Khan, T.B. Shaik, M. Ashraf, Bioorg. Med. Chem. Lett. 20 (2010) 5229-5231; (e) B.V. Subba Reddy, N. Rajeswari, M. Sarangapani, et al., Bioorg. Med. Chem. Lett. 22 (2012) 2460-2463; (f) M. Shiri, M.A. Zolfigol, H.G. Kruger, Z. Tanbakouchian, Chem. Rev. 110 (2010) 2250-2293. |
| [3] |
(a) G. Gao, Y. Han, Z.H. Zhang, ChemistrySelect 2 (2017) 11561-11564; (b) J. Xiao, H. Wen, L. Wang, et al., Green Chem. 18 (2016) 1032-1037. |
| [4] |
(a) K. Fuji, Chem. Rev. 93 (1993) 2037-2066; (b) I. Denissova, L. Barriault, Tetrahedron 59 (2003) 10105-10146; (c) K.W. Quasdorf, L.E. Overman, Nature 516 (2014) 181-191; (d) R. Long, J. Huang, J. Gong, Z. Yang, Nat. Prod. Rep. 32 (2015) 1584-1601; (e) X.P. Zeng, Z.Y. Cao, Y.H. Wang, F. Zhou, J. Zhou, Chem. Rev. 116 (2016) 7330-7396; (f) J. Feng, M. Holmes, M.J. Krische, Chem. Rev. 117 (2017) 12564-12580; (g) Y. Li, S. Xu, Chem. Eur. J. 24 (2018) 16218-16245; (h) C. Li, S.S. Ragab, G. Liu, W. Tang, Nat. Prod. Rep. 37 (2020) 276-292. |
| [5] |
(a) M. Delgado-Rebollo, A. Prieto, P.J. Pérez, ChemCatChem 6 (2014) 2047-2052; (b) M.B. Johansen, M.A. Kerr, Org. Lett. 12 (2010) 4956-4959; (c) A. DeAngelis, V.W. Shurtleff, O. Dmitrenko, J.M. Fox, J. Am. Chem. Soc. 133 (2011) 1650-1653; (d) X. Gao, B. Wu, W.X. Huang, M.W. Chen, Y.G. Zhou, Angew. Chem. Int. Ed. 54 (2015) 11956-11960; (e) V. Arredondo, S.C. Hiew, E.S. Gutman, I.D.U.A. Premachandra, D.L. Van Vranken, Angew. Chem. Int. Ed. 56 (2017) 4156-4159. |
| [6] |
Z. Du, Y. Xing, P. Lu, Y. Wang, Org. Lett. 17 (2015) 1192-1195. DOI:10.1021/acs.orglett.5b00089 |
| [7] |
R.R. Singh, R.S. Liu, Chem. Commun. 53 (2017) 4593-4596. DOI:10.1039/C7CC01304C |
| [8] |
X. Hu, F. Chen, Y. Deng, H. Jiang, W. Zeng, Org. Lett. 20 (2018) 6140-6143. DOI:10.1021/acs.orglett.8b02613 |
| [9] |
(a) C.J. Li, Chem. Rev. 105 (2005) 3095-3166; (b) C.J. Li, L. Chen, Chem. Soc. Rev. 35 (2006) 68-82; (c) A. Chanda, V.V. Fokin, Chem. Rev. 109 (2009) 725-748; (d) R.N. Butler, A.G. Coyne, Chem. Rev. 110 (2010) 6302-6337; (e) M.B. Gawande, V.D.B. Bonifácio, R. Luque, P.S. Branco, R.S. Varma, Chem. Soc. Rev. 42 (2013) 5522-5551. |
| [10] |
(a) R. Breslow, Acc. Chem. Res. 24 (1991) 159-164; (b) R. Breslow, Acc. Chem. Res. 37 (2004) 471-478; (c) S. Narayan, J. Muldoon, M.G. Finn, et al., Angew. Chem. Int. Ed. 44 (2005) 3275-3279; (d) W. Li, G. Yin, L. Huang, et al., Green Chem. 18 (2016) 4879-4883; (e) C. Wu, X. Xin, Z.M. Fu, et al., Green Chem. 19 (2017) 1983-1989; (f) S. Peng, Y.X. Song, J.Y. He, et al., Chin. Chem. Lett. 30 (2019) 2287-2290; (g) Y. Kim, C.J. Li, Green Synth. Catal. 1 (2020) 1-11. |
| [11] |
(a) J.P. Bégué, D. Bonnet-Delpon, B. Crousse, Synlett (2004) 18-29; (b) I.A. Shuklov, N.V. Dubrovina, A. Börner, Synthesis (2007) 2925-2943; (c) I. Colomer, A.E.R. Chamberlain, M.B. Haughey, T.J. Donohoe, Nat. Rev. Chem. 1 (2017) 0088; (d) J. Wencel-Delord, F. Colobert, Org. Chem. Front. 3 (2016) 394-400. |
| [12] |
(a) H.F. Motiwala, R.H. Vekariya, J. Aubé, Org. Lett. 17 (2015) 5484-5487; (b) R.H. Vekariya, J. Aubé, Org. Lett. 18 (2016) 3534-3537; (c) G.X. Li, J. Qu, Chem. Commun. 46 (2010) 2653-2655; (d) M. De Rosa, A. Soriente, Eur. J. Org. Chem. (2010) 1029-1032; (e) Z. Sadiq, M. Iqbal, E.A. Hussain, S. Naz, J. Mol. Liq. 255 (2018) 26-42; (f) W.C. Shiyao Lu, Qilong Shen, Chin. Chem. Lett. 30 (2019) 2279-2281. |
| [13] |
M.B. Rubin, R. Gleiter, Chem. Rev. 100 (2000) 1121-1164. DOI:10.1021/cr960079j |
| [14] |
A. Saba, Synth. Commun. 24 (1994) 695-699. DOI:10.1080/00397919408012648 |
| [15] |
Y. Yu, Q. Sha, H. Cui, K.S. Chandler, M.P. Doyle, Org. Lett. 20 (2018) 776-779. DOI:10.1021/acs.orglett.7b03912 |
| [16] |
(a) R.D.C. Gallo, A.C.B. Burtoloso, Green Chem. 20 (2018) 4547-4556; (b) H.H. San, S.J. Wang, M. Jiang, X.Y. Tang, Org. Lett. 20 (2018) 4672-4676. |
| [17] |
(a) S.F. Zhu, C. Chen, Y. Cai, Q.L. Zhou, Angew. Chem. Int. Ed. 47 (2008) 932-934; (b) S.F. Zhu, Y. Cai, H.X. Mao, J.H. Xie, Q.L. Zhou, Nat. Chem. 2 (2010) 546-551. |
| [18] |
X.J. Lv, Y.H. Chen, Y.K. Liu, Org. Lett. 21 (2019) 190-195. DOI:10.1021/acs.orglett.8b03654 |
| [19] |
B.M. Trost, S. Malhotra, P. Koschker, P. Ellerbrock, J. Am. Chem. Soc. 134 (2012) 2075-2084. DOI:10.1021/ja206995s |
| [20] |
(a) D. Vuluga, J. Legros, B. Crousse, et al., J. Org. Chem. 76 (2011) 1126-1133; (b) S. Khaksar, S.M. Vahdat, M. Gholizadeh, S.M. Talesh, J. Fluorine Chem. 136 (2012) 8-11; (c) C. Pi, X. Yin, X. Cui, Y. Ma, Y. Wu, Org. Lett. 21 (2019) 2081-2084. |