 2021, Vol. 32
2021, Vol. 32 


Anion recognition is an important research topic in chemistry due to the key roles of anions in chemical, biological, material, and environmental processes [1-4]. The Hofmeister effect, proposed by Franz Hofmeister in 1888 [5], refers to the action of certain salts to decrease the solubility of proteins while others increase it. Numerous studies have reported the "Hofmeister series" of anions: F–, SO42–, CH3COO–, Cl–, Br–, NO3–, I–, ClO4−, SCN−, BF4–, and PF6– [6, 7]. The former species are referred to as kosmotropes (salting-out anions), while the latter are called chaotropes (salting-in anions). These terms were also originally used to describe the capacity of a particular anion to "make" or "break" the water structure [8], respectively. In other words, kosmotropic anions exhibit a strong hydration capacity with hydrophilicity, and chaotropic anions exhibit a weak hydration capacity with hydrophobicity [9]. This was thought to be central to the mechanism of the Hofmeister effect. However, this idea has been challenged by the phenomenon of anionic chaotropes that increase the solubility of proteins and unfold their tertiary structure, resulting in the molten globule state in water [10, 11]. For example, it was found that the molten globule state is generally favored in the presence of anionic chaotropes because the large and well-organized hydrophobic surfaces maximize ion interactions [12-14]. These studies revealed the possibility that anions interact directly with the hydrophobic surfaces of molecules [15, 16].
In 2011, Gibb et al. provided unequivocal evidence that chaotropic anions exhibit an affinity to a hydrophobic cavity [17]. In 2018, the same group used NMR spectra to further confirm the hydrophobicity of chaotropic anions in the Hofmeister series by using a synthetic positive-charge-appended macrocyclic host in water at a salt concentration of ~2 mmol/L [7]. This is because in some instances, a reverse Hofmeister effect is observed at different salt concentrations: e.g., lysozyme exhibited a direct Hofmeister effect at a high ionic strength, but the reverse Hofmeister effect appeared at low salt concentrations [18]. Therefore, it is believed that examining the Hofmeister effect at a lower salt concentration such as at micromolar levels can lead to a greater understanding of this phenomenon. Here, taking advantage of the high sensitivity of fluorescence methods that enable the tracking of molecular interactions at nanomolar concentrations, we attempt to identify the subtly hydrophobic properties of chaotropic anions at very low concentrations (5.0–100.0 μmol/L), which can be distinguished by a tunable hydrophobic effect from a cationic fluorophore-based aggregation in an aqueous solution via the visualization of fluorescent signals.
The unique property of aggregation-induced emission (AIE) offers a new mechanism for fluorophore design [19]. Generally, AIE fluorophores are non- or weakly fluorescent in good solvents, but they emit a strong fluorescence upon aggregation in poor solvents. In most cases, the obtained AIE entities are usually attributed to the intrinsic hydrophobic effect of the constituent monomer in water [20]. Therefore, a more strongly hydrophobic effect is expected in the aggregate state. However, researchers have focused on the unique photophysical property of AIE, while neglecting to investigate and exploit the hydrophobic effect in the AIE system. Herein, we studied the hydrophobic effect and identified that the AIE-triggered tunable hydrophobic effect can be utilized to systematically study the hydrophobic properties of chaotropic anions in the Hofmeister series for the first time.
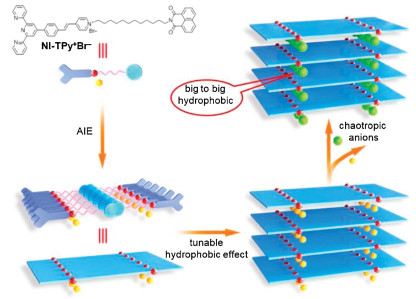
Scheme 1 shows a π-chromophore-containing amphiphile (NI-TPy+Br–) as the AIE monomer, which contains naphthalimide (NI) at one end of a flexible aliphatic chain, a terpyridine skeleton, and a styrylpyridinium cation (TPy+) as the polar segment at the other end (details in Supporting information). This AIE monomer assembled into nanofibers in an aqueous solution and emitted two independent AIE signals at 398 nm and 545 nm, respectively. Most importantly, it was expected that the alternate self-aggregation of NI [21] leads to the formation of a small hydrophobic cavity for chaotropic-anion competitive binding. For example, after the addition of iodide anions to the NI-TPy+Br– aggregation solution, it was found that the fluorescence emission at 545 nm of the aggregates was completely quenched, whereas no obvious emission change was observed at 398 nm. Most interestingly, the amplification of hydrophobicity could be significantly tuned by controlling the aggregates and concentrations of NI-TPy+ in water, which thus provide a tunable positive-charge-appended hydrophobic effect to match the suitable chaotropic anions with a distinct fluorescent signal.

|
Download:
|
| Scheme 1. Chemical structure of NI-TPy+Br–, and the related schematic representation of AIE-triggered tunable hydrophobic effect and anion recognition with the Hofmeister series in water. | |
{kind=link}
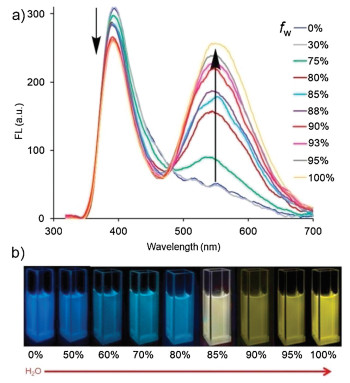
As a general protocol, the AIE behavior of NI-TPy+Br– was first evaluated in CH3CH2OH/water mixtures with different water fractions (fw), which enabled fine-tuning of the solvent polarity and the extent of solute aggregation. As show in Fig. 1, the pure CH3CH2OH solution of NI-TPy+Br– shows blue fluorescence with an emission maximum around 400 nm. With the gradual addition of water into CH3CH2OH (fw = 70 vol%), no obvious change occurs in the blue emission of NI-TPy+Br– and a new bathochromically shift emission is visible around 545 nm, which can be attributed to the inhibited twisted intramolecular charge transfer (TICT) of TPy+ with the increasing solvent polarity [22]. The emission peak at 545 nm is dramatically enhanced with the continued increase in fw, and a typical AIE effect is observed. However, the emission intensity at 400 nm is just slightly weakened. This may be ascribed to the fluorescence emission balance between the decrease monomer emission intensity at 450 nm of TPy+ (Fig. S1 in Supporting information) and the increase π-π stacking emission intensity at 380 nm of NI (the efficiency of the intersystem crossing process of NI decreased with increasing solvent polarity, thus leading to the increasing fluorescence efficiency) [21, 23] in the NI-TPy+Br– aggregation state. In addition, as shown in Fig. S2 (Supporting information), the monomer of NI and TPy+ exhibit similar absorption peaks around 350 nm in the UV–vis spectra, indicating that both fluorophores can be independently excited by the same wavelength. Thus, the results of this study demonstrated a unique AIE system that can be constructed using two independent fluorophores via the strategy of "single excitation, multiple emissions".

|
Download:
|
| Fig. 1. (a) Fluorescence spectra of NI-TPy+Br– (10.0 μmol/L) in CH3CH2OH with increasing water fractions (H2O/CH3CH2OH, v/v), λex = 345 nm. (b) Photographs of solution of NI-TPy+Br– in CH3CH2OH with increasing water fractions (H2O/CH3CH2OH, v/v) under UV light at 365 nm. | |
{kind=link}
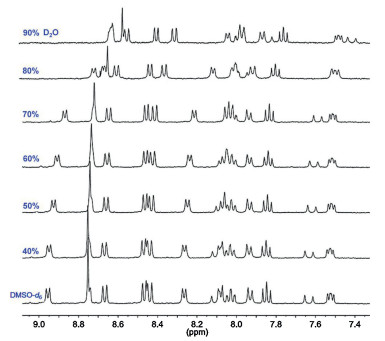
The aggregation behaviors of NI-TPy+Br– were further studied by carrying out fluorescence measurements of the aqueous solutions of NI-TPy+Br– with different concentrations. The critical aggregation concentration (CAC) of the AIE monomers was determined to be 2.6×10−6 mol/L (Fig. S3 in Supporting information). The Tyndall effect was observed for the aqueous solutions of NI-TPy+Br– at concentrations higher than the CAC (Fig. S4 in Supporting information), indicating the existence of aggregates. Most interestingly, the obviously increased upfield proton shift in the 1H NMR spectra of NI-TPy+Br– in DMSO-d6 with the increased water (D2O) fractions indicates the strengthening of the hydrophobic effect in the aggregates (Fig. 2). Furthermore, the 1H NMR spectra of NI-TPy+Br– at different concentrations in DMSO-d6 with the same water fractions (Fig. S5 in Supporting information) also revealed that the aggregation-induced hydrophobic effect can be tuned by modifying the NI-TPy+Br– concentrations. Specifically, a higher concentration results in a stronger hydrophobic effect in a polar solvent. In addition, SEM images indicate that the aggregation of NI-TPy+Br– resulted in a ribbon-like self-assembled structure (Fig. S6 in Supporting information), and atomic force microscopy (AFM) image suggested that the height of the aggregates up to 18 nm (Fig. S7 in Supporting information).

|
Download:
|
| Fig. 2. 1H NMR spectra of NI-TPy+Br– (1.0mmol/L) in DMSO-d6 with increasing water (D2O) fractions (D2O/DMSO-d6, v/v). | |
{kind=link}
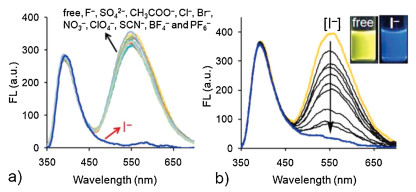
Upon addition of various anions (sodium or potassium salt) to the aqueous solution of NI-TPy+Br− (10.0 μmol/L), it was observed that only I– anion caused a remarkable fluorescence changes (Fig. 3a). Fig. 3b shows the changes in the fluorescence spectra of NI-TPy+Br– in an aqueous solution upon the addition of increasing concentrations of I– (NaI, from 0 to 10.0 μmol/L). It can be seen that the fluorescence intensity of NI-TPy+Br– at 545 nm significantly decreased, while that at 400 nm remained almost unchanged. DLS data suggested that the aggregate size changed from 300 nm to 400 nm in solution (Fig. S8 in Supporting information). Therefore, the decreased fluorescence intensity at 545 nm can be attributed to the heavy atom effect of I– to the TPy+ fluorophore rather than the disaggregation of NI-TPy+Br–. The linear relationship between the fluorescence intensity changes at 545 nm and the amount of anions added revealed that the detection limit of (LOD) the aggregation-based probe for I– was 9.5×10–8 mol/L (Fig. S9 in Supporting information). Furthermore, an estimation of the interference of other co-existing anions in the selective response of NI-TPy+Br– to I– was studied. The fluorescence intensity was almost identical to that obtained in the absence of the other species, indicating that the NI-TPy+Br– aggregates in water were highly selective and sensitive toward I– at this concentration (Fig. S10 in Supporting information).

|
Download:
|
| Fig. 3. (a) Response emission intensities of NI-TPy+Br– (10.0 μmol/L) for various anions (each of 20 μmol/L) in water. (b) Fluorescence emission changes of NI-TPy+Br– (10.0 μmol/L) with various concentrations of NaI (0–10 μmol/L) in water, λex = 345 nm. | |
{kind=link}
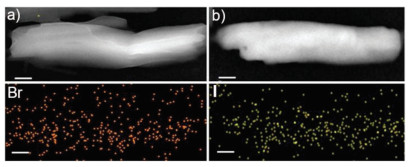
TEM images of NI-TPy+Br– in water after the addition of I– show a morphology similar to the ribbon-like morphology of NI-TPy+Br– aggregates (Fig. 4), and the EDS results show that the aggregate blocks were composed of C, N, O, and I. This indicates that I– as the counter anion substituted for Br– in the NI-TPy+-based aggregate entities without changing their assembly frameworks, which was also supported by the unchanged fluorescence emission at 398 nm.

|
Download:
|
| Fig. 4. TEM images of (a) NI-TPy+Br–, (b) NI-TPy+I–, and the corresponding EDS elemental mapping images. Scale bar is 100 nm. | |
{kind=link}
Further, we speculate that the hydrophobic effect of the NI-TPy+Br– aggregates is the driving force for I– to compete with Br–, which was supported by the response of NI-TPy+Br– in organic solvents to I–. As show in Fig. S11 (Supporting information), NI-TPy+Br– exhibited blue monomer emission in DMSO or CH3CH2OH solution, indicating that no aggregation of NI-TPy+Br– occurred because of its good solubility in the solvents. Upon the addition of I– to the solution, no obvious fluorescence spectral changes was observed, which verified that the hydrophobic effect resulting from the aggregation of NI-TPy+Br– in water is the crucial factor driving the competition between I– and Br–.
It is well known that anion hydration as being strongly dependent upon the surface charge density and progressing from strong hydration for small ions of high charge density (kosmotropes) to weak hydration for large ions with low charge density (chaotropes) [24]. In particular, according to Collins's report, oppositely charged anions and cations with a similar hydration capacity combine to form strong ion pairs, whereas weak unstable ion pairs are formed if the oppositely charged anions and cations have a large difference in the hydration capacity in water; this is also called "small to small, big to big" [25]. Therefore, it is reasonable that in the aggregation assemblies, NI-TPy+—a big cationic entity—will tend to match with the hydrophobic anion I– that is bigger than Br– based on the Hofmeister series. In other words, the assembly of NI-TPy+I– is ascribed to have the tendency of "big hydrophobic to big hydrophobic" (Scheme 1).
In this work, we discovered that the amplification of hydrophobicity by aggregation was significantly dependent upon the aggregates and concentrations of NI-TPy+Br– in water (Fig. 2). This result indicates the potential to gain more detailed information regarding the relationship between the tunable hydrophobicity of the cationic fluorophore-based aggregates and chaotropic anions. As shown in Fig. S12 (Supporting information), upon the addition of different chaotropic anions to the aqueous solution of NI-TPy+Br– at different concentrations, it was seen that the presence of I– induced a completely quenched emission at 545 nm, and no significant spectral changes were observed for other anions, when the concentration of NI-TPy+Br– was between 5.0μmol/L and 10.0 μmol/L. However, as the concentration of NI-TPy+Br– was increased to 50 μmol/L and then to 100 μmol/L, it was found that quenched emission tendency at 545 nm was obviously tardiness and a moderate decrease in the intensity in the presence of I– ions was observed. This result indicated that the selective binding ability of the aggregates with I– was inhibited by the increased concentration of NI-TPy+Br– due to the dependence of the hydrophobic effect on the concentration.
In contrast, a continuous enhancement of the fluorescence emission at 545 nm was observed in the presence of ClO4– and BF4– with increasing NI-TPy+Br– concentration in water. As shown in Fig. S13 (Supporting information), a significant enhancement was observed when the concentration of NI-TPy+Br– was fixed at 50 μmol/L for ClO4– and 100 μmol/L for BF4–. The enhanced fluorescence emission can be attributed to the fact that ClO4– or BF4– as the counter anion makes the NI-TPy+ moiety adopt a more rigid molecular planarity and thus enhance the AIE intensity. These results imply that the hydrophobic effect in the case of 50 μmol/L NI-TPy+Br– tends to match the ClO4– anion. Similarly, 100 μmol/L NI-TPy+Br– in water exhibited the most obvious fluorescence response signal for BF4–, and this is attributed to the substantially stronger hydrophobic effect expected to be generated in this case, resulting in the tendency to match larger hydrophobic BF4− anions. The corresponding TEM images indicate that the ribbon-like morphology was maintained in the aggregates after the addition of these anions, and the EDS results show the presence of Cl (Fig. S14 in Supporting information), B, and F atoms (Fig. S15 in Supporting information) in the aggregate blocks, respectively.
Notably, no fluorescence response was observed in the presence of PF6–, which exhibits the largest hydrophobic effect in chaotropic anions according to the Hofmeister series, irrespective of the NI-TPy+Br– concentration. We hypothesized that this may be attributed to the fact that the hydrophobic effect resulting from the aggregation of NI-TPy+Br– may not be sufficient to match the hydrophobic effect of PF6–. We wanted to continue increasing the concentration of NI-TPy+, but the limited solubility of NI-TPy+Br– in water made this difficult. Overall, the above observations successfully support the phenomenon that the big hydrophobic cationic aggregates match the big hydrophobic chaotropic anions, and this is in agreement with the Hofmeister series.
In summary, an unprecedented tunable hydrophobic effect was discovered in a cationic fluorophore-based aggregation assembly with the AIE property, and this was exploited to evaluate the anion binding behaviors in aqueous solutions with the Hofmeister effect. Due to the unique AIE signals of the cationic fluorophore, the binding between the cationic entities with AIE and the anions can be subtly monitored in real-time on the basis of the fluorescence spectra. The results demonstrated that the tunable hydrophobic effect derived from the self-assembly of small organic cationic molecule in water plays a key role in the selective binding of chaotropic anions with the Hofmeister series in line with the tendency "big hydrophobic to big hydrophobic." We hope this work will facilitate the further construction of supramolecular assemblies for applications in fields such as anion sensing, separation, and new AIE materials.
Declaration of competing interestThere are no conflicts to declare.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21871063) and Guizhou University (No. YJSCXJH(2019)012).
| [1] |
J.L. Sessler, P.A. Gale, W.S. Cho, Anion Receptor Chemistry, RSC Publishing, Cambridge, 2006.
|
| [2] |
K. Brak, E. Jacobsen, Angew. Chem. Int. Ed. 52 (2013) 534-561. DOI:10.1002/anie.201205449 |
| [3] |
M. Jaspers, A.E. Rowan, P.H.J. Kouwer, Adv. Funct. Mater. 25 (2015) 6503-6510. DOI:10.1002/adfm.201502241 |
| [4] |
V. Etacheri, R. Marom, R. Elazari, et al., Energy Environ. Sci. 4 (2011) 3243-3262. DOI:10.1039/c1ee01598b |
| [5] |
F. Hofmeister, Naunyn-Schmiedeberg's Arch. Pharmacol. 24 (1888) 247-260. DOI:10.1007/BF01918191 |
| [6] |
P.L. Nostro, B.W. Ninham, Chem. Rev. 112 (2012) 2286-2322. DOI:10.1021/cr200271j |
| [7] |
J.H. Jordan, C.L.D. Gibb, A. Wishard, et al., J. Am. Chem. Soc. 140 (2018) 4092-4099. DOI:10.1021/jacs.8b00196 |
| [8] |
B. Hribar, N.T. Southall, V. Vlachy, et al., J. Am. Chem. Soc. 124 (2002) 12302-12311. DOI:10.1021/ja026014h |
| [9] |
A.S. Thomas, A.H. Elcock, J. Am. Chem. Soc. 129 (2007) 14887-14898. DOI:10.1021/ja073097z |
| [10] |
D. Hamada, S.I. Kidokoro, H. Fukada, et al., Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 10325-10329. DOI:10.1073/pnas.91.22.10325 |
| [11] |
R.M. Lynn, Y. Konishi, H.A. Scheraga, Biochemistry 23 (1984) 2470-2477. DOI:10.1021/bi00306a023 |
| [12] |
Y. Zhang, P.S. Cremer, Curr. Opin. Chem. Biol. 10 (2006) 658-663. DOI:10.1016/j.cbpa.2006.09.020 |
| [13] |
Y.J. Zhang, P.S. Cremer, Annu. Rev. Phys. Chem. 61 (2010) 63-83. DOI:10.1146/annurev.physchem.59.032607.093635 |
| [14] |
R.L. Baldwin, Biophys. J. 71 (1996) 2056-2063. DOI:10.1016/S0006-3495(96)79404-3 |
| [15] |
L.M. Pegram, T. Wendorffa, R. Erdmanna, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 7716-7721. DOI:10.1073/pnas.0913376107 |
| [16] |
L.M. Pegram, M.T.J.J. Record, Phys. Chem. B 111 (2007) 5411-5417. DOI:10.1021/jp070245z |
| [17] |
C.L.D. Gibb, B.C. Gibb, J. Am. Chem. Soc. 133 (2011) 7344-7347. DOI:10.1021/ja202308n |
| [18] |
Y. Zhang, P.S. Cremer, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 15249-15253. DOI:10.1073/pnas.0907616106 |
| [19] |
(a) D. Ding, K. Li, B. Liu, B.Z. Tang, Acc. Chem. Res. 46 (2013) 2441-2453; (b) T. Xiao, H. Wu, G. Sun, et al., Chem. Commun. 56 (2020) 12021-12024; (c) T. Xiao, W. Zhong, L. Zhou, et al., Chin. Chem. Lett. 30 (2019) 31-36; (d) T. Xiao, J. Wang, Y. Shen, et al., Chin. Chem. Lett. (2020), doi: http://dx.doi.org/http://dx.doi.org/10.1016/j.cclet.2020.10.037 |
| [20] |
(a) J. Mei, N.L.C. Leung, R.T.K. Kwok, et al., Chem. Rev. 115 (2015) 11718-11940; (b) L. Shao, J. Sun, B. Hua, F. Huang, Chem. Commun. 54 (2018) 4866-4869; (c) X. Yan, H. Wang, C.E. Hauke, et al., J. Am. Chem. Soc. 137 (2015) 15276-15286. |
| [21] |
P. Gopikrishna, N. Meher, P.K. Iyer, ACS Appl. Mater. Interfaces 10 (2018) 12081-12111. DOI:10.1021/acsami.7b14473 |
| [22] |
G. Zhou, X. Zhang, X.L. Ni, J. Hazard. Mater. 384 (2020) 121474. DOI:10.1016/j.jhazmat.2019.121474 |
| [23] |
V. Wintgens, P. Valet, J. Kossanyi, et al., Faraday Trans. 90 (1994) 411-421. DOI:10.1039/FT9949000411 |
| [24] |
K.D. Collins, Biophys. J. 72 (1997) 65-76. DOI:10.1016/S0006-3495(97)78647-8 |
| [25] |
K.D. Collins, Methods 34 (2004) 300-311. DOI:10.1016/j.ymeth.2004.03.021 |