 2021, Vol. 32
2021, Vol. 32 


b Department of Chemistry, Graduate School of Science, and Integrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo Chikusa, Nagoya 464-8602, Japan;
c Institute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo Chikusa, Nagoya 464-8602, Japan;
d New York University Abu Dhabi, PO Box 129188, Abu Dhabi, United Arab Emirates
Solid-state luminescent materials have been always a desirable cutting-edge research field on account of the potential values in optoelectronic technologies [1-8]. The emergence of triplet-involved emitters thrives many advanced material fields such as electroluminescence, molecular sensors, and time-resolved bioimaging [9-11]. Emission mechanism of these materials involves triplet state so that quantum efficiency could surpass the limitation of the efficiency of traditional fluorescent materials and/or materials with afterglow emission which is used to exclude the influence of fluorescence in some fields [12-14]. How to design triplet-involved materials is always an important subject in materials research. The design method for constructing Thermally Activated Delayed Fluorescence (TADF) had already been proposed by reducing the energy gap between singlet and triplet states [15]. However, the suitable design approaches for Room-Temperature Phosphorescent (RTP) materials are still under developing.
Initially, the construction of RTP materials has been largely focused on metal-containing inorganic materials on the basis of transition metal complexes [16, 17], because metal atoms could promote the spin–orbit coupling (SOC) and boost the intersystem crossing (ISC) [18]. In 2011, Kim et al. designed a doped organic crystal with persistent RTP characteristic, demonstrating the importance of heavy atom effect in the generation of RTP [19]. The proposal of crystalline-induced phosphorescence (CIP) made an enormous splash on single molecular RTP materials, indicating the imporance of crystalline state [20, 21]. From then on, several types of crystal structures, including H-aggregation [22-24], intermolecular electronic coupling [25], π…π interactions [26, 27], halogen bonds [28], and so on [29, 30], have been proposed for illustrating the mechanism of RTP. Even though, the known mechanisms for triplet-involved emitters are still underdeveloped. Further investigations on the mechanism of triple-invovled materials are crucial for molecular design and the fabrication of materials.
Most of RTP organic crystalline materials could be assigned to crystallization-induced emission, namely emission by the restriction of intramolecular motions (RIM) [20, 21]. It means emission behavior originates from the limitation of molecular rotation and vibration. Molecules could rotate and vibrate freely in solution, where non-radiative transitions are usually dominant. However, in a rigid environment (for example, frozen solution), molecules cannot rotate and vibrate freely, leading to RTP emission. In this work, we propose a new concept of excited-state conformation capture to construct crystallization-induced emission RTP materials which are different from the aforementioned RTP materials. The optimum excited states of monomers in solution or rigid enviornment have totally distinct conformations when compared with those in crystal structure. A simple restriction of molecular movements is not enough to catch the optimum excited-state conformations with bright RTP emissions. However, because of the strong interactions in crystalline state, excited-state structures would be changed, forming new optimum excited-state conformations with RTP emission behaviors. In other words, supramolecular interactions could capture new optimum excited-state conformations for bright RTP emissions in crystal. The design concept, emission behavior, and intermolecular interactions will be discussed in this work.
Herein, we reported a series of molecules lack of emissions in solution and rigid environment but have different triplet-involved emission behaviors in crystalline forms. This article will be divided into four steps to demonstrate our diverse design concepts on the basis of excited-state conformation capture. Two crystalline-induced TADF molecules are designed and synthesized (step 1). Those molecules could form supramoelecular chains by strong hydrogen bonds which could capture excited-state conformations. Starting with those chains, efficient RTP (step 2), heavy-atom-free RTP (step 3) and ultra RTP materials (step 4) are attained by seperating aggregate effectsand ehnhancing ISC process. Our work harvests the triplet energies of pure organic materials efficiently by capturing excited-state structures, and investigates the essence of partial crystalline-induced triplet-involved emitters in-depth.
Step 1: Excited-state conformation capture for TADF.
Compounds 1a and 1b were obtained by one step synthesis (details see Supporting information) [31]. As shown in Fig. 1a, the crystals 1a and 1b demonstrate bright emissions with peak maxima at 427 and 463 nm, respectively. The decay spectra further revealed that the crystals have long-lived luminescent behaviors besides prompt luminescence (Figs. S1a–d in Supporting information). In order to confirm the origin of such emission component, temperature-dependent decay spectroscopy (Figs. 1b and c) was performed. The ratio of delayed component increased with the temperature rising from 250 K to 330 K, which determined the behavior of thermal activation energy for TADF. For further investigation of TADF emission behavior, monomer's photophysical properties of 1 were employed. It should be noted that 1a and 1b are all non-emissive in deoxygenated solution, on the plates of thin-layer chromatography (TLC plate, aggregate state), and on the spin-coated films (Figs. S2‒S4 in Supporting information), which is totally different from tranditional TADF emitters reported in the literatures, indicating the existance of triplet-involved crystallization-induced emission (CIE) behavior. In general, the solutions of triplet-involved molecules emit phosphorescence under frozen conditions on account of RIM [20, 21]. In contrast, at 77 K, the solutions of 1a and 1b could not emit luminescence (Fig. S5 in Supporting information). Such crucial difference reveals that molecular structures and the rigidity of environment are not the most important factors for the TADF properties and there must be some other impacts in crystal structures which may be momentous for the observed TADF behavior.

|
Download:
|
| Fig. 1. Emission spectra of crystals 1, insert: photograph of 1a (upper) and 1b (lower) under 365 UV light (a); temperature-dependent decay spectra of crystals 1a (b) and 1b (c) measured at 420 and 460 nm, respectively; adiabatic ΔEst between optimum singlet excited state and optimum triplet excited state in crystal structure (d); excited-state conformations of 1a and 2b calculated in THF (e); excited-state conformation (red) were calculated by freezing the upper and lower molecules (green) based on S1 state (f). | |
{kind=link}
For the sake of investigation on such significant emission between monomer and crystal, single-crystal X-ray diffraction analysis was performed on 1a and 1b. Both crystals adopt the same crystal systems (monoclinic) and space groups (P21/c). Molecules 1a and 1b take molecular conformations with dihedral angles of 76.66° and 64.45° between the central phenyl ring and the fluorophenyl ring, respectively (Figs. S7a and b in Supporting information).
Each molecule in both 1a and 1b connects others through four robust N-H…O-C interactions (2.10 Å for 1a and 2.27 Å for 1b), forming infinite supramolecular chains (Figs. 2a and b). These chains connect with other chains through weak C-H…π (2.60 Å for 1a and 2.65 Å for 1b) interactions (Figs. S7c and d in Supporting information). In the structures of these two crystals, the main packing modes are supramolecular chains formed by hydrogen bonds. This unique chain-like structure may be essential for triplet-involved emission.

|
Download:
|
| Fig. 2. Supramolecular chains of 1a (a), 1b (b), forming by hydrogen bonds. Packing modes of 2a (c), and 2b (d). | |
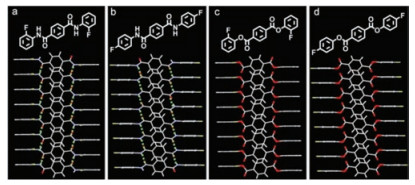{kind=link}
To verify that supramolecular chain is the key factor of triplet-involved emission, compounds 2a and 2b were synthesized in which two NH moieties were replaced with O atoms to eliminate the supramolecular chains. The monomers of 2 could not emit luminescence in solution, on the plates of thin-layer chromatography (TLC plate, aggregate state), and on the spin-coated films, even at a extreme low temperature (77 K) (Figs. S2‒S5). Interestingly, in crystalline forms, the crystals 2 still could not emit luminescence (Fig. S8 in Supporting information), ensuring that the rigidity of crystals is not the main reason for TADF emission behavior of 1. For a further comparison, single-crystal X-ray diffractions of 2 were investigated in comparison with those of 1. For 2, they inherit similar chain-like packing modes, but without intermolecular interactions in chains (Figs. 2c and d). The lack of intermolecular interactions in packing modes prevents the formation of supramolecular chains. Considering such important differences in the crystal structures of 1 and 2, it could be concluded that the unjoined structures of 2a and 2b handicaps the formation of triplet-involved emissions, and that the formation of supramolecular chains must be crucial for triplet-involved emissions.
The above experimental results proved the key role of supramolecular chains in emission behaviour. However, two problems still exist: (1) How do supramolecular chains influence the emission behavior between monomers (in solution or in rigid environment) and molecules in crystal? (2) What is the origin of TADF emission?
In order to solve these two problems, theoretical calculations using time-dependent density functional theory (TD-DFT) in solution and crystal state were performed by Gaussian 16 program [32]. Taking 1a and 2b as examples (Fig. 1e), in solution, excited-state conformations are significantly twisted because of the lack of sufficient constraint in molecular conformation, leading to a decreation of molecular energy and ensuring a spin-forbidden transition process (oscillator strength of single state=0). Thus such twist excited-state structure leads to a non-radiative transition process which prohibits the emission of monomers. In order to investigate emission behaviors in crystal state, a segment of a supramoleculat chain with three molecules were employed to provide similar crystal environment in which upper and lower molecules are frozen to provide interactions, while the central molecule could act freely (Fig. 1f). The calculation results were all without imaginary frequencies which means that real local minima were reached. In crystal structure, molecules 1a locked by strong hydrogrn bonds in supramolecular chain could not rotate or vibrate frequently, forming a new type of exicted state conformation. In crystals 2, molecules could tranform to the twisting conformation as in solution because of the absence of interactions in packing mode, though crystal has a rigid enviroment. Such evidences proved that the supramolecular chains influence emission behaviors by capturing a new optimum excited state conformation, which leads to the emission in crystalline form.
As for the origin of TADF, the adiabatic energy gap in crystal structure between the optimum singlet excited state and the optimum triplet exicited state was calculated to be 0.4 eV, which is suitable for TADF emission (Fig. 1d) [15]. Simultaneously, the energy gap could be reduced with the extension of chain lengths based on the crystal structure (Table S2 in Supporting information).
In this part, a new optimum excited-state conformation was obtained by using supramolecular chains as a driving force of excited-state conformation capture technique. In order to utilize such method sufficiently, we tend to design materials with RTP emission and investigate the mechanism of RTP.
Step 2: Spin-orbit coupling enhancement for RTP.
TADF emitters could be required by excited-state conformation capture. How to foster RTP materials is the main topic in this step. Heavy atoms (for example, Br atom) could enhance SOC and accelerate ISC process between singlet and triplet states, thus they were constantly selected to attain RTP materials [33, 34]. Similarly, compound 3 was designed by introducing two Br atoms on the central phenyl ring to enhance SOC process. Compound 3 does not emit in frozen solution (77 K), on TLC plate, and on spin-coating film (Figs. S3‒S5), implying that the molecular structure and the rigidity of environment are not the most crucial factors for emission. However, the crystals obtained by sublimation demenstrate green emission. The emission maximum was located at 501 nm as shown in Fig. 3a. Temperature-dependent decay spectra indicated that the emission of 3 could be assigned to RTP (Fig. 3b).

|
Download:
|
| Fig. 3. Emission spectrum of crystal 3, insert: photograph of 3 under 365 UV light (a); temperature-dependent decay spectra of 3 (b); energy level diagram and the corresponding SOC constants of the monomer of 3 (c); crystal structure of 3′ (d); supramolecular chain of 3 (e); excited-state conformation (red color) was calculated by freezing the upper and lower molecules (green color) based on T1 state (f); NTO isosurfaces of the trimer of 3 with different transition types (g). | |
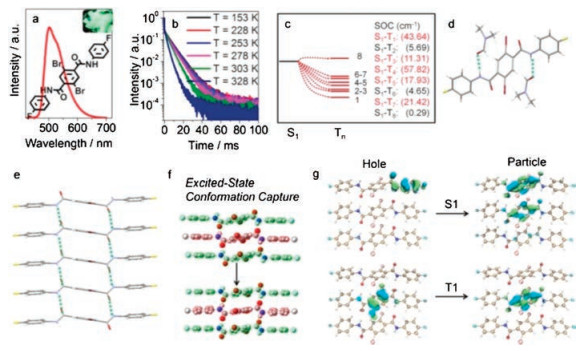{kind=link}
The space group of 3 is P-1, confirmed by Single crystal X-ray diffraction analysis. Each molecule connecting with others through four C=O…H-N hydrogen bonds (2.08 Å) forms the supramolecular chains (Fig. 3e). The Br atoms in one molecule could provide efficient external heavy atom effects due to the appropriate distances (3.66 Å) and tremendous overlap between Br atoms in one molecule and phenyl rings in other molecules (Fig. S10 in Supporting information).
Additionally, a pseudo-polymorph (3′) of compound 3 was obtained. Crystal 3′ was grown in DMF at a chilling temperature. In contrast to the bright emission of crystal 3, 3′ is non-emissive under UV irradiation though it possesses suitable crystalline morphology (Fig. S11 in Supporting information). The unit cell of 3′ contains one molecule 3 and one DMF molecule. The isolated molecule forms N-H…O-C interactions (2.09 Å) with two other DMF molecules, prohibiting the construction of supramolecular chains (Fig. 3d). The hydrogen bonds, which are formed with DMF molecules, could not lock excited-state conformation, resulting in free rotation and vibration of molecule 3. Such results further proved the importance of supramolecular chain and its capability in excited-state conformations capturing.
The theoretical calculations of 3 were carried out in the same way with 1a and 2b. Single molecule was calculated by TD-DFT and trimer state was calculated by UDFT method. In solution, the excited-state conformation is also a twisting structure with the oscillator strength of single state=0 (Fig. S9b in Supporting information). However, in crystalline state, excited-state conformation could be captured by supramolecular chains as an optimum structure with radiative transition (Fig. 3f). Excited-state conformation capture method constitutes the cornerstone of RTP emission behavior of 3. For a further investigation of the difference of emission behavior between 1 and 3, natural transition oribital (NTO) analysis was performed. As shown in Fig. 3g, Figs. S9c and S9d (Supporting information), singlet excited-state transition forms of 1 and 3 both possess aggregate effect while triplet excited-state transition forms are both attributed to the influence of monomers whose conformations are captured by supramolecular chains. Similar transitions mean transition forms are not the momentous factors for RTP emissions. Hence, distinct emission behavior could be assigned to the existence of Br atoms. Br atoms could enhance ISC process, which is proved by the large SOC constants shown in Fig. 3c and Fig. S13a (Supporting information). SOC constants were calculated by ORCA [35, 36], and corresponding diagrams were drawn by VMD and Multiwfn [37, 38]. Most of the SOC constants of monomers are larger than those of trimers, indicating that RTP should be mainly attributed to the influence of monomers. Br atoms could facilitate the RTP emissions by reducing reverse intersystem crossing (RISC) process as well as accelerating ISC process.
Based on the NTOs results, it could be proposed that TADF emissions may originate from aggregate state while RTP emissions may derive from the monomer's excited-state conformation captured by supramolecular chains. Therefore, in the next step, we manage to weaken the hydrogen bonds in supramolecular chains to impair the status of aggregate state and the construction of heavy-atom-free RTP materials.
Step 3: Elimination of aggregate effect for heavy-atom-free RTP.
To obtain efficient and heavy-atom-free RTP materials, the influence of aggregate effect is intended to undermine and thus the effect of monomer's excited-state conformation capture would be enhanced. The hydrogen atoms bonding to the nitrogen atoms are replaced with phenyl rings in order to reduce the aggregate state. This design concept would enlarge the distance of two neighbouring molecules, leads to the weakening of intermolecular-aggregation effect. Simultaneouly, the extension of intermolecular distances may impair the hydrogen bonds strength, which may also handicap the transition process between molecules. Based on these considerations, compound 4 was designed and synthesized.
The emission behavior of 4 in solution is identical to compouds 1–3. Compound 4 does not emit in solution (77 K), on TLC plate, and on spin-coating film (Figs. S2‒S5). In crystalline state, 4 exhibits intensive emission in air with the peak maxima at 512 nm (Fig. 4a). Temperature-dependent decay spectra from 77 K to 328 K confirmed that the emission of 4 is undoubtedly ascribed to RTP (Fig. 4b). Space group of compound 4 is P-1. As shown in Fig. 4d, although H atoms in N-H moieties are replaced with phenyl rings, packing mode is quite similar to those of 1–3 (molecules are locked by four hydrogen bonds). Each molecule connects with adjacent molecules through four C=O…H hydrogen bonds, by which supramolecular chains are formed. The phenyl rings forming hydrogen bonds with carbonyl groups are almost parallel to the hydrogen bonds, which indicates that the replacement with phenyl groups does not disturb the formation of supramolecular chains. Compared with those of 1–3, the length of hydrogen bonds of 4 increases to 2.71 Å, indicating an extension of intermolecular distance. Both of the experimental results and single crystal structure analysis prove the reliability of our hyothesis that TADF emissions originate from aggregate states while RTP emissions derive from the monomer's excited-state conformation captured by supramolecular chains.

|
Download:
|
| Fig. 4. Emission spectrum of crystal 4. Insert: photograph of 4 under 365 UV light (a); temperature-dependent decay spectra of 4 (b); energy level diagram and the corresponding SOC constants of the monomer of 4 (c); supramolecular chain of 4 (d); excited-state conformation (red color) was calculated by freezing the upper and lower molecules (green color) based on T1 state (e); NTO isosurfaces of the trimer of 3 with different transition types (f). | |
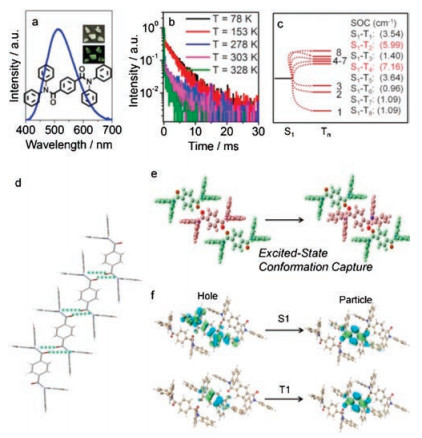{kind=link}
Theoretical calculations were carried out in the same way of 3. In crystalline state (Fig. 4e), the excited-state conformation could still be captured by strong hydrogen bonds in supramolecular chains, forming the cornerstone of triplet-involved emissions. To investigate the origin of RTP, ISC process was investigated at first. As can be seen in SOC constants (Fig. 4c), compared with 3 (Fig. 3c), SOC process is not enhanced dramatically, indicating that RTP could be caused by other reasons. NTOs indicate that the transition types both of singlet excited state and triplet excited state convert to monomer's forms, demonstrating the weakening of aggregate state which is caused by the extension of intermolecular distances and the reduction of hydrogen bonds (Fig. 4f). Besides, the energy gaps between singlet and triplet state are 0.62 eV for monomer and 0.68 eV for trimer, respectively, which prohibit RISC process. All above mentioned facts contribute in the promoting of the RTP emission.
It is concluded from this step that RTP emission originates from monomer's excited-state confomration capture. In the next step, we intend to design ultralong RTP by summarising the concepts of Steps 1–3.
Step 4: Stabilization of triplet state for URTP.
In Step 1, we figured out that excited-state conformation could be captured by supramolecular chains, and constructs triplet-involved materials. In Step 2, the introduction of Br atoms enhances spin-orbit coupling and facilitates the ISC process. In Step 3, heavy-atom-free RTP material was obtained by eliminating aggregate effect and achieving monomer's excited-state confomration capture. Based on these results, we tend to design ultralong RTP (URTP) materials by using other interactions as implements for supramolecular chains.
Four conditions should be satisfied in order to obtain URTP materials by supramolecular chains: (1) excited-state conformation capture, (2) monomer's transition formation, (3) enhancement of ISC process, and (4) stability of triplet excitions. Hence, π…π stacking which is widely used to stablise triplet excitions [22, 23] and Br atom which is usually introduced for an enhancement of ISC process [33-35] were employed in the construction of URTP material. In this step, π…π stacking could also serve as an implement of excited-state conformation capture for the formation of supramolecular chains. At the same time, it was found that the range of π…π stacking is around 3.50 Å (face to face), which could undermine molecular aggregation. Therefore, compound 5 was designed and synthesized.
Crystals of compound 5 were obtained with good crystalline morphology from methanol/DCM solution. In crystalline state, compound 5 displays fluorescence peaking at 430 nm and excellent RTP with the emission maximum at 500 nm (Fig. 5a). The decay spectrum of RTP component is shown in Fig. 5b, and the lifetime of 5 was 99 ms, which is indicative of a good performance of RTP characteristic. In order to find the origin of RTP emission, the emission behavior of monomers was investigated. The solution of 5 at room temperature is non-emissive (Fig. S2). However, under frozen solution, phophorescent emission could be achieved, though intensity is feebly detected by naked eyes (Fig. S6b in Supporting information). Such weak emission with peak maximum around 500 nm proved that the origin of RTP should be attributed to the influence of monomers, but such restriction of molecular movements is not enough for RTP emission as in crystals. The interactions in crystals must play key role in the RTP behavior.

|
Download:
|
| Fig. 5. Emission spectra of crystals 5, insert: photograph of 5 under daylight and with afterglow (a); decay spectrum of 5 (b); energy level diagram and the corresponding SOC constants of the monomer of 5 (c); time-resolved indentification pattern of 5 (d); supramolecular chain of 5 (e); excited-state conformation (red color) was calculated by freezing the upper and lower molecules (green color) based on T1 state (f); NTO isosurfaces of the trimer of 5 with different transition types (g). | |
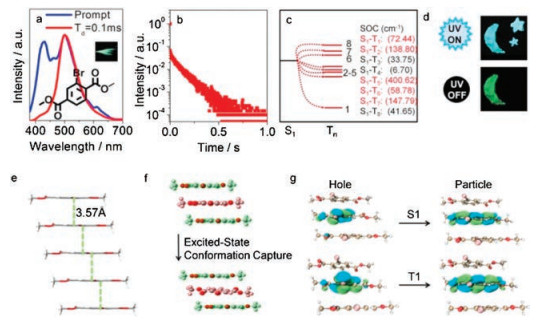{kind=link}
In the crystal structure, the unit cell possesses two molecules. These two molecules form supramolecular chains through π…π packing. The distances between adjacent phenyl rings are 3.57 Å (Fig. 5e) and 3.62 Å (Fig. S12 in Supporting information), and the centroid-centroid distances of adjacent phenyl rings are both 3.98 Å. It could be concluded that, compared with the hydrogen bonds in 1‒4, π…π stacking interaction increases the distances among molecules, which reduce the influence of aggregate effects. Besides, the existence of π…π stacking may not only capture a new optimum excited-state conformation but also stabilize triplet excitions while Br atom could benefit the ISC process, both of which are responsible for the URTP. Such crystal structure and emission under frozen conditions preliminarily proved the efficacy of our design methods mentioned previously.
Theoretical calculation were performed on one of the conformations in crystal form as a further evidence. The excited-state conformation is captured by π…π stacking (Fig. 5f), while Br atom benefits the ISC process as shown by SOC constants in Fig. 5c. NTO analysis (Fig. 5g) showed that, because of the extension of intermolecular distances by π…π stacking, transition forms of both singlet and triplet excited states are located on a monomer, eliminating aggregate effect.
In the summary of this step, (1) the introducation of π…π stacking, which is an implement for excited-state conformation capture in this step, could more effectively restrict the movements of molecules than a simple rigid environment for RTP emission; (2) π…π stacking eliminates aggregate effect by extending the distance among molecules, which may hinder the RISC process, and stabilizes triplet excitons; (3) Br atom benefits spin-orbit coupling and enhance intersystem crossing process. It could be concluded that RTP emission behavior should originate from monomer's excited-state conformation capture.
The feature of URTP is persistent emission after the removal of excitation sources. Consequently, the URTP materials generally function as color-encoding or secure patterns. As shown in Fig. 5d, we demonstrated the potential application of 5 in time-resolved identification as an illustration. The pattern was consisted of two components. The moon was constructed by 5 with URTP and the stars were comprised of 1a. The moon and stars displayed cyan emission under 365 UV light. While only the moon exhibited pure green emission after the removal of UV light and the emission could be detected by naked eyes, indicating a marvelous application in secure protections.
In this work, a new concept of excited-state conformation capture by supramolecular chains was proposed for the explanation of mechanism of triplet-involved emissions and for the construction of different types of triplet-involved emitters (TADF, RTP, heavy-atom-free RTP, and URTP). First of all, we designed two molecules with crystalline-induced TADF. It could be summarised that, different from the optimum excited-state structure in solution, excited-state conformation in crystal state is captured by supramolecular chain, forming a new optimum sturcture with bright emission. Next, bright RTP emitter was designed by introducing Br atoms based on the similar supramolecular chains. The introduction of Br atoms enhances spin-orbit coulping and improves ISC process. From a comparison of TADF and RTP emitters, it could be proposed that TADF may originate from aggregate effect. Then aggregate effect was eradicated by extending the distances between molecules in supramolecular chains for heavy-atom-free RTP. Finally, π…π stacking was employed as an interaction in supramolecular chains for excited-state conformation capture and for the elimination of aggregate effects, constructing URTP materials. There methods not only enrich the mechanism of triplet-involved materials but also provide potient ways, which intramolecular interactions are used to capture excited-state conformations, for constructing amorphous phosphorescent organic materials. Further investigations are undergoing.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentThis work was supported by the National Natural Science Foundation of China (No. 51773077).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.013.
| [1] |
Z. Fu, K. Wang, B. Zou, Chin. Chem. Lett. 30 (2019) 1883-1894. DOI:10.1016/j.cclet.2019.08.041 |
| [2] |
H. Liu, Y. Wang, H. Zhang, et al., Chem. Sci. 10 (2019) 227-232. DOI:10.1039/C8SC03135E |
| [3] |
I.O. Koshevoy, Y.C. Chang, P.T. Chou, et al., J. Am. Chem. Soc. 134 (2012) 6564-6567. DOI:10.1021/ja3018994 |
| [4] |
D. Li, H. Zhang, Y. Wang, Chem. Soc. Rev. 42 (2013) 8416-8433. DOI:10.1039/c3cs60170f |
| [5] |
S. Tan, X. Wu, Y. Zheng, Y. Wang, Chin. Chem. Lett. 30 (2019) 1951-1954. DOI:10.1016/j.cclet.2019.08.003 |
| [6] |
D. Zhou, C.H. Ryoo, D. Liu, et al., Adv. Opt. Mater. 8 (2019) 1901021. |
| [7] |
Y. Liu, Z. Yin, X. Wang, et al., J. Mater. Chem. C 8 (2020) 8971-8979. DOI:10.1039/D0TC01612H |
| [8] |
D. Zhou, D. Liu, G. Xie, et al., ACS Appl. Mater. Interfaces 11 (2019) 24339-24348. DOI:10.1021/acsami.9b07511 |
| [9] |
L. Zhang, Y.F. Wang, C.F. Chen, et al., Chin. Chem. Lett. 32 (2021) 740-744. DOI:10.1016/j.cclet.2020.07.041 |
| [10] |
H.T. Feng, S. Zou, B.Z. Tang, et al., J. Am. Chem. Soc. 142 (2020) 11442-11450. DOI:10.1021/jacs.0c02434 |
| [11] |
L. Huang, Y. Luo, G. Zhang, et al., Angew. Chem. Int. Ed. 130 (2018) 16278-16282. DOI:10.1002/ange.201808861 |
| [12] |
Q. Lin, Z. Li, Q. Yuan, Chin. Chem. Lett. 30 (2019) 1547-1556. DOI:10.1016/j.cclet.2019.06.016 |
| [13] |
H. Uoyama, K. Goushi, C. Adachi, et al., Nature 492 (2012) 234-238. DOI:10.1038/nature11687 |
| [14] |
Z. Yang, Z. Chi, M.P. Aldred, et al., Chem. Soc. Rev. 46 (2017) 915-1016. DOI:10.1039/C6CS00368K |
| [15] |
Q. Zhang, B. Li, C. Adachi, et al., Nat. Photon. 8 (2014) 326-332. DOI:10.1038/nphoton.2014.12 |
| [16] |
G. Li, C. Adachi, Y.B. Dong, et al., Chin. Chem. Lett. 30 (2019) 1931-1934. DOI:10.1016/j.cclet.2019.08.006 |
| [17] |
A. Obolda, M. Zhang, F. Li, Chin. Chem. Lett. 27 (2016) 1345-1349. DOI:10.1016/j.cclet.2016.06.030 |
| [18] |
K. Van den Eeckhout, P.F. Smet, D. Poelman, Materials 3 (2010) 2536-2566. DOI:10.3390/ma3042536 |
| [19] |
O. Bolton, K. Lee, J. Kim, et al., Nat. Chem. 3 (2011) 205-210. DOI:10.1038/nchem.984 |
| [20] |
W. Yuan, X. Shen, B.Z. Tang, et al., J. Phys. Chem. C 114 (2010) 6090-6099. DOI:10.1021/jp909388y |
| [21] |
Y. Gong, L. Zhao, B.Z. Tang, et al., Chem. Sci. 6 (2015) 4438-4444. DOI:10.1039/C5SC00253B |
| [22] |
E. Lucenti, A. Forni, E. Cariati, et al., J. Phys. Chem. Lett. 8 (2017) 1894-1898. DOI:10.1021/acs.jpclett.7b00503 |
| [23] |
Z. An, X. Liu, W. Huang, et al., Nat. Mater. 14 (2015) 685-690. DOI:10.1038/nmat4259 |
| [24] |
E. Lucenti, A. Forni, E. Cariati, et al., Angew. Chem. Int. Ed. 56 (2017) 16302-16307. DOI:10.1002/anie.201710279 |
| [25] |
Z. Yang, Z. Chi, M.R. Bryce, et al., Angew. Chem. Int. Ed. 55 (2016) 2181-2185. DOI:10.1002/anie.201509224 |
| [26] |
J. Yang, X. Zhen, Z. Li, et al., Nat. Commun. 9 (2018) 840. DOI:10.1038/s41467-018-03236-6 |
| [27] |
Y. Wen, H. Liu, B. Yang, et al., J. Mater. Chem. C 7 (2019) 12502-12508. DOI:10.1039/C9TC04580E |
| [28] |
W. Wang, Y. Zhang, W.J. Jin, Coord. Chem. Rev. 404 (2020) 213107. DOI:10.1016/j.ccr.2019.213107 |
| [29] |
Z. He, W. Li, W.Z. Yuan, et al., Chin. Chem. Lett. 30 (2019) 933-936. DOI:10.1016/j.cclet.2019.03.015 |
| [30] |
H. Bhatia, I. Bhattacharjee, D. Ray, J. Phys. Chem. Lett. 9 (2018) 3808-3813. DOI:10.1021/acs.jpclett.8b01551 |
| [31] |
N. Cheng, Q. Yan, D. Zhao, et al., Cryst. Eng. Comm. 16 (2014) 4265-4273. DOI:10.1039/C4CE00089G |
| [32] |
M. Frisch, G. Trucks, H. Schlegel, et al., Gaussian 16, Revision B.01. Wallingford CT: Gaussian, Inc., 2016.
|
| [33] |
H. Shi, W. Huang, Y. Zhao, et al., Cryst. Growth Des. 16 (2016) 808-813. DOI:10.1021/acs.cgd.5b01400 |
| [34] |
Z. Wang, T. Li, X. Ma, et al., Chin. Chem. Lett. 31 (2020) 2929-2932. DOI:10.1016/j.cclet.2020.05.015 |
| [35] |
F. Weigend, Phys. Chem. Chem. Phys. 8 (2006) 1057-1065. DOI:10.1039/b515623h |
| [36] |
F. Neese, WIREs Comput. Mol. Sci. 8 (2017) e1327. |
| [37] |
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580-592. DOI:10.1002/jcc.22885 |
| [38] |
W. Humphrey, A. Dalke, K. Schulten, J. Mol. Graphics 14 (1996) 33-38. DOI:10.1016/0263-7855(96)00018-5 |