 2021, Vol. 32
2021, Vol. 32 


b Key Laboratory of Molecular Virology and Immunology, Institut Pasteur of Shanghai, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200031, China;
c College of Pharmacy and Chemistry, Dali University, Dali 671000, China;
d Frontiers Science Center for Materiobiology and Dynamic Chemistry, East China University of Science and Technology, Shanghai 200237, China;
e School of Life Science and Technology, ShanghaiTech University, Shanghai 201210, China
Despite decades of effort to control the disease, malaria is still one of the most threatening problems in global public health governance. According to the statistical data of World Health Organization (WHO) in 2018, malaria has caused 228 million clinical episodes and 405, 000 deaths, among them, Plasmodium falciparum (P. falciparum) induced the highest mortality [1]. Since no malarial vaccines have been approved clinically, chemotherapies are still the mainstays of treating and preventing malaria [2, 3]. Unfortunately, the emergence and widespread of multi-drug resistant malaria has severely emasculated the therapeutic efficacy of several early-approved antimalarial agents [4-6]. Especially the prevalence of resistance against artemisinin-based combination therapies (ACTs) has significantly raised concerns about the prospect of malaria control [7-11]. Therefore, discovery of next-generation antimalarial drugs with novel therapeutic targets to overcome the obstacles of drug resistance is an important and urgent task for malaria elimination.
Histone deacetylase (HDAC), selectively hydrolyzing acetyl groups in the amino acid residues of histone, is a critical post-translational modulator. Currently, five P. falciparum HDACs (PfHDACs) have been identified which are divided into Zn2+-containing class I (PfHDAC1) and class Ⅱ (PfHDAC2/3) HDACs, and NAD+-containing class Ⅲ (PfSir2A/2B) HDACs [12]. SincePfHDACs play important roles in regulating the acetylation levels of both histone and non-histone malarial proteins, which affect the survival and reproduction of parasite, these enzymes are considered as promising antimalarial drug targets [12-15]. Merck Research Laboratory reported the pioneering PfHDAC inhibitor apicidin in 1996 [16], and several groups subsequently published their researches of PfHDAC inhibitor as potential antimalarial agents 12-15]. However, in spite of more than two decades of research, PfHDAC inhibitors never successfully entered clinical treatment or trail. Although several HDAC inhibitors displayed antimalarial potency, their toxicity has become the major fault of further development [12]. Hence developments of antimalarial HDAC inhibitor in the future should focus on enhancing the therapeutic safety of novel compounds.
In previous work, we discovered that the clinical antitumor HDAC inhibitor Quisinostat (1) was a promising antimalarial agent with potent PfHDAC1 inhibition, but the severe toxic effects limited its further application [17, 18]. Previous work also explored the structure–activity relationship (SAR) of Quisinostat, and revealed that replacing the 4-aminomethyl piperidine linker with other diamine linkers could significantly regulate the bioactive properties [18]. In order to reduce the toxicity and discover new chemical prototype of antimalarial agent, we envisaged a novel series ofPfHDAC inhibitors based on displacing the original diamine linker by a spirocyclic 2, 6-diazaspiro[3.3]heptane linker (Scheme 1). Modification of the CAP fragment was then carried out to jointly improve the safety. Therefore, 30 novel spirocyclic linker derivatives with various CAP groups were designed and synthesized, and their antimalarial activities and cytotoxicity were systematically evaluated in this work.

|
Download:
|
| Scheme 1. Chemical modification strategy of novel PfHDAC inhibitors and their synthetic routes. Reagents and conditions: (a) DIPEA, DCM, 25 ℃; (b) HCl/1, 4-dioxane (4 mol/L), DCM, 25 ℃; (c) R-CHO, HOAc, NaBH(OAc)3, DCE, 25 ℃; (d) K2CO3, MeOH, H2O, 65–70 ℃; (e) THPONH2, EDCI, HOBt, TEA, DMF, 25 ℃; (f) HCl/1, 4-dioxane (4 mol/L), DCM, 25 ℃. | |
The general synthetic routes of compounds 2–31 were depicted in Scheme 1 [19]. Briefly, 2, 6-diazaspiro[3.3]heptane-2-carboxylic acid tert-butyl ester half oxalate (32) underwent nucleophilic substitution reaction with ethyl 2-chloropyrimidine-5-carboxylate under basic condition to produce intermediate 33. The tert-butoxycarbonyl (Boc) group in 33 was removed under acidic condition and the resultant free amine 34 underwent reductive amination to produce 35–64. After basic hydrolysis of ethyl ester in the 35–64, the corresponding acid underwent condensation with O-(tetrahydro-2H-pyran-2-yl)hydroxylamine to produce 65–94. Finally compounds 2–31 were prepared by removing the 2-tetrahydropyranyl (THP) group in 65–94 under treatment with organic hydrogen chloride solution.
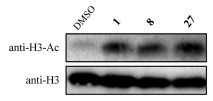
In vitro plasmodium-killing assay of compounds 2–31 against drug-sensitive (3D7) and chloroquine-resistant (Dd2) P. falciparum strains, and their cytotoxicity against two human cell lines (HepG2 and 293T) were systematically evaluated with Quisinostat and dihydroartemisinin (DHA) as positive controls. Analysis of the data summarized in Table 1 revealed some noteworthy observations from the SAR study of compounds 1–31: (1) diamine group displacement could slightly attenuate antimalarial potency and significantly improve the selectivity (1 vs. 2); (2) small monocyclic and large aromatic CAP groups could retain or attenuate potency, but the selectivity was not improved or even greatly reduced (3–6 and 25–26 vs. 2); (3) bicyclic CAP groups, such as naphthalyl, quinolyl, isoquinolyl, piperonyl, dihydrobenzofuryl and other azaindolyl groups were disadvantageous to potency, while benzofuryl, benzothiophenyl and indazolyl groups slightly enhanced potency although the selectivity was compromised (10–24 vs. 2); (4) the N-methyl group in the indolyl CAP groups was critical to potency and 2-indolyl group was superior in improving both potency and selectivity (7–9 vs. 2); (5) introducing methoxyl group to 3-indolyl group undermined the potency, while introducing cyano and halogen groups could retain or enhance potency (27–31 vs. 2). Encouragingly, the two most potent compounds 8 and 27 also significantly inhibited three multi-drug resistant P. falciparum strains (Table S2 in Supporting information) [20]. Further investigations revealed that the metabolic stability of 8 and 27 was enhanced since their half-life (T1/2) toward mouse liver microsomes in vitro was longer than that of 1 (Table 2). Western blot assay also displayed that 8 or 27 could also induce hyperacetylation of parasite histone H3 compared with the blank group, which indicated that 8 and 27 were PfHDAC inhibitors the same as compound 1 (Fig. 1).
|
|
Table 1 In vitro antimalarial activity and cytotoxicity of compounds 2–31. |
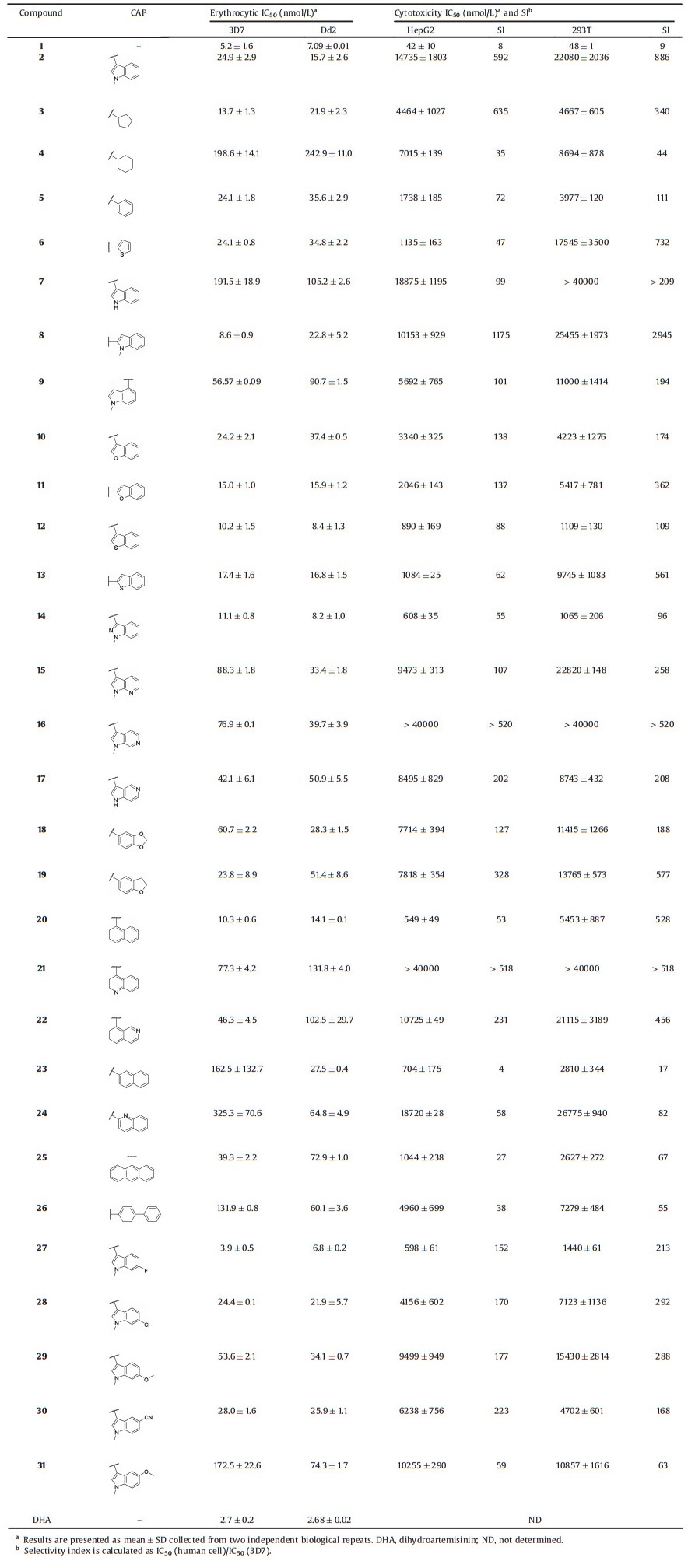
|
|
Table 2 In vitro metabolic stability of compounds 1, 8 and 27. |
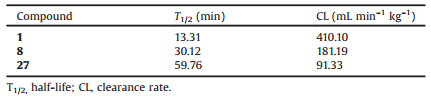
{kind=link}

|
Download:
|
| Fig. 1. Hyperacetylation analysis of P. falciparum histone H3 by compounds 1, 8 and 27. | |
{kind=link}
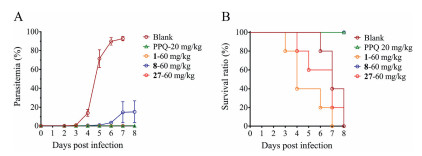
Given the remarkable in vitro antimalarial potency of compounds 8 and 27 against multiple P. falciparum strains along with alleviative cytotoxicity and stable metabolic properties, we intended to evaluate their in vivo blood-stage antimalarial activities. Since P. falciparum cannot directly infect rodent animal model [21], we selected a widely-accepted rodent P. yoelii malaria infection model with piperaquine phosphate (PPQ) as positive control [20, 22]. The animal experiment was approved by Animal Welfare and Committee of Institut Pasteur of Shanghai, Chinese Academy of Sciences (IACUC Issue No. A2018009). Female BALB/c mice were randomly divided into 5 groups and each one was inoculated with 1×105 parasites on day 0. Subsequently, tested drug solution or solvent alone (for blank group) were administered intraperitoneally once daily since day 1 to day 5. As shown in Fig. 2, the mice in blank group finally died from the hyperparasitemia, and the mice in compound 1 administration group died quickly from the compound toxicity. Compound 27 displayed strong in vivo efficacy in killing malaria parasites, but there was still a problem of toxicity. Although compound 8 was unable to eliminate the parasites thoroughly, it still delayed the progress of disease and exhibited the inhibition rate of 84% on day 8 while resulting in no mouse death. Therefore, both compounds 8 and 27 inhibited the in vivo parasite growth and compound 8 further displayed significantly reduced toxicity compared to compound 1.

|
Download:
|
| Fig. 2. In vivo erythrocytic antimalarial activity of compounds 1, 8 and 27. Percentage of parasitemia (A) and survival ratio (B) of compounds 1, 8 and 27 at doses of 60 mg/kg. Blank is the solvent-treated group. Each group had five mice. Piperaquine phosphate (PPQ) was the positive control. Tail vein blood smears were prepared on the indicated days, and the percentage of parasitemia was determined via microscopic examination of 5000 red bold cells and presented as mean±SD. | |
{kind=link}
In summary, based on the structure of the lead compound 1, we designed and synthesized 30 novel spirocyclic linker derivatives (2–31), and their antimalarial activity and cytotoxicity were systematically evaluated. An explicit SAR was acquired and revealed that introduction of the 2, 6-diazaspiro[3.3]heptane linker could remarkably reduce cytotoxicity, and modifications of CAP fragment could further attenuate cytotoxicity and regulate antimalarial activity. Among all the derivatives, compounds 8 and 27 displayed potent antimalarial activity with half maximal inhibitory concentration (IC50) values below 10 nmol/L. These compounds also exhibited strong killing efficacy against several multi-drug resistant clinical P. falciparum isolates, and showed enhanced metabolic stability against mouse liver microsomes in vitro compared to 1. Western blot analysis demonstrated that compounds 8 and 27 are PfHDAC inhibitors the same as lead compound 1. Moreover, both compounds 8 and 27 showed killing efficacy against parasites in animal experiment, and the toxicity of compound 8 in animals was significantly improved compared with 1. These results implied that the risks of potential toxic effects could be reduced by reasonable structural modification. Overall, 8 and 27 had the potential to be new lead compounds for further development.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Key R&D Program of China (Nos. 2017YFB0202600, 2018YFA0507300), the National Mega-project for Innovative Drugs of China (No. 2019ZX09721001- 004-003), the National Natural Science Foundation of China (Nos. 81872747, 81903457, 31972169), the National Science and Technology Major Project (No. 2018ZX10101004003001), the Innovative Research Team of High-level Local Universities in Shanghai, the National Special Fund for State Key Laboratory of Bioreactor Engineering (No. 2060204), the Shanghai Sailing Program (No. 19YF1412600) and the Shanghai Morning Light Program (No. 18CG33).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.12.023.
| [1] |
World Health Organization, World Malaria Report, 2019. https://apps.who.int/iris/handle/10665/330011.
|
| [2] |
J.N. Burrows, E. Burlot, B. Campo, et al., Parasitology 141 (2014) 128-139. DOI:10.1017/S0031182013000826 |
| [3] |
World Health Organization, Guidelines for the Treatment of Malaria, 3rd. ed., (2015). https://www.who.int/docs/default-source/documents/publications/gmp/guidelines-for-the-treatment-of-malaria-eng.pdf.
|
| [4] |
I.B. Müller, J.E. Hyde, Future Microbiol. 5 (2010) 1857-1873. DOI:10.2217/fmb.10.136 |
| [5] |
B. Blasco, D. Leroy, D.A. Fidock, Nat. Med. 23 (2017) 917-928. DOI:10.1038/nm.4381 |
| [6] |
D. Menard, A. Dondorp, Cold Spring Harb. Perspect. Med. 7 (2017) a025619. DOI:10.1101/cshperspect.a025619 |
| [7] |
L. Zhu, J. Tripathi, F.M. Rocamora, et al., Nat. Commun. 9 (2018) 5158. DOI:10.1038/s41467-018-07588-x |
| [8] |
J. Wang, C. Xu, F.L. Liao, et al., N. Engl. J. Med. 380 (2019) 2087-2089. DOI:10.1056/NEJMp1901233 |
| [9] |
A.M. Talman, J. Clain, R. Duval, R. Ménard, F. Ariey, Trends Parasitol. 35 (2019) 953-963. DOI:10.1016/j.pt.2019.09.005 |
| [10] |
World Health Organization, Status Report on Artemisinin Resistance and ACT Efficacy, December, 209. https://www.who.int/docs/default-source/documents/publications/gmp/who-cds-gmp-2019-17-eng.pdf.
|
| [11] |
A. Uwimana, E. Legrand, B.H. Stokes, et al., Nat. Med. 26 (2020) 1602-1608. DOI:10.1038/s41591-020-1005-2 |
| [12] |
G.S. Hailu, D. Robaa, M. Forgione, et al., J. Med. Chem. 60 (2017) 4780-4804. DOI:10.1021/acs.jmedchem.6b01595 |
| [13] |
K.T. Andrews, A. Haque, M.K. Jones, Immunol. Cell Biol. 90 (2012) 66-77. DOI:10.1038/icb.2011.97 |
| [14] |
K.T. Andrews, T.N. Tran, D.P. Fairlie, Curr. Pharm. Design 18 (2012) 3467-3479. |
| [15] |
R. Fioravanti, N. Mautone, A. Rovere, D. Rotili, A. Mai, Curr. Opin. Chem. Biol. 57 (2020) 65-74. DOI:10.1016/j.cbpa.2020.05.008 |
| [16] |
S.J. Darkin-Rattray, A.M. Gurnett, R.W. Myers, et al., Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 13143-13147. DOI:10.1073/pnas.93.23.13143 |
| [17] |
L.B. Jiang, Z.H. Huang, WO2017143964, 2017.
|
| [18] |
Z.H. Huang, R.X. Li, T.K. Tang, et al., Cell Discov. (2020). DOI:10.1038/s41421-020-00215-4 |
| [19] |
J.W.J. Dickens, I.N. Houpis, Y.L. Lang, et al., WO2008138918 November 20, (2008).
|
| [20] |
Y. Yang, Y. Yu, X. Li, et al., J. Med. Chem. 60 (2017) 1994-2005. DOI:10.1021/acs.jmedchem.6b01733 |
| [21] |
M.B. Jimenez-Diaz, T. Mulet, S. Viera, et al., Antimicrob. Agents Chemother. 53 (2009) 4533-4536. DOI:10.1128/AAC.00519-09 |
| [22] |
T. Ono, T. Tadakuma, A. Rodriguez, Exp. Parasitol. 115 (2007) 310-313. DOI:10.1016/j.exppara.2006.09.008 |