 2021, Vol. 32
2021, Vol. 32 


b School of Clinical Medicine, Chengdu University of TCM, Chengdu 610072, China;
c Department of Emergency, Binzhou Medical University Hospital, Binzhou 256603, China;
d Unité de Biologie Fonctionnelle et Adaptative, Université de Paris, Paris 75013, France;
e Department of Pharmacy, Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, Personalized Drug Therapy Key Laboratory of Sichuan Province, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610072, China;
f Department of EICU, Sichuan Academy of Medical Sciences & Sichuan Provincial People's Hospital, Chengdu 610072, China
According to statistics, about 80%–85% of lung cancers are non-small cell lung cancer (NSCLC). Squamous cell carcinoma, adenocarcinoma and large cell carcinoma are the main subtypes of NSCLC. The small cell lung cancer (SCLC) accounts for 10%–15% of the diagnosed lung cancer cases in clinic [1]. Unfortunately, the SCLC metastasizes faster than NSCLC. Studies showed the Src homology region 2 (SH2)-containing protein tyrosine phosphatase 2 (SHP2) mutations occur frequently in different types of leukemia, only 2% mutation in lung cancers behind colon and endometrial cancers [2-4]. However, the immunohistochemical analysis on the lung tumor biopsy samples obtained from clinic showed 58% of which are SHP2 mutants positive [5]. Obviously SHP2 mutations account for a high proportion in lung cancer. Studies also showed that the activation of the SHP2 cancelled drug resist in the Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS)-mutant NSCLC cells [6]. In addition, the E76K mutant SHP2 induces lung tumorigenesis is confirmed in transgenic mice while saving the expression of the functional SHP2 suppresses the NSCLC progressing in the same transgenic mice model [7].
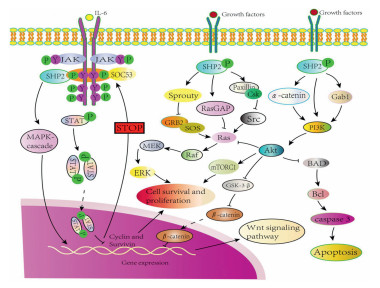
The phosphatases play critical roles in modulating the proteins' biological functions. Specifically, the phosphatases correspond to protein kinases, forming a "two-way switch" in which phosphorylation and dephosphorylation dynamically regulates the target protein activity [8, 9]. Since the enzymes participate in gene transcription and protein translation are also controlled by this "two-way switch" system, the phosphatases-kinases system decides the basic biological functions like cell proliferation and metabolism [10, 11]. Non-receptor protein tyrosine phosphatases (NRPTPs) exhibit striking structural diversity, which enables binding to specific proteins or recognizing specific subcellular locations [12]. SHP2 is a member of NRPTPs family encoded by the tyrosine-protein phosphatase non-receptor type 11 (PTPN11) [13]. The SHP2 is involved in multiple cell signaling transduction pathways including promoting the activation of RAS/mitogen-activated protein kinase (MAPK), RAS/extracellular-signal-regulated kinase (ERK), phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT), Janus kinase/signal transducers and activators of transcription (JAK/STAT) and Hippo/yes-associated protein (YAP) pathways. Besides, SHP2 also modulates the expression of growth factors, cytokines and hormones [14-16]. In the SHP2-deficient bone marrow macrophages, the induction of macrophage colony-stimulating factor (M-CSF), a secreted cytokine which causes hematopoietic stem cells to differentiate into macrophages or other related cell type enhanced the PI3K/AKT activation. Inhibition of SHP2 upregulates JAK/STAT dephosphorylation and is related with Hippo pathway which are known as tumor suppressors, negatively control YAP function in Pca cell line (prostate cancer cell) [17-19]. Otherwise, SHP2 in epidermal growth factor receptor (EGFR) mutant lung cancer enables active YAP/TAZ through RAS/MAPK signaling axis [20]. The recent studies demonstrated that SHP2 has a wide range of network cascading effects. Specifically, evidences showed that SHP2 modulates cell death pathway programmed death protein 1/programmed death ligand 1 (PD-1/PD-L1) and acts as a B/T lymphocyte attenuator (BTLA) which attenuates the immune evasion. The above findings indicate SHP2 maybe a potential new target for cancer treatment [21, 22]. The Fig. 1 summarizes the currently known SHP2 signaling pathways.

|
Download:
|
| Fig. 1. SHP2 associated signaling pathway in this review. | |
The crystal structures of SHP2 from Michael J. Eck group showed that SHP2 contains two tandem SH2 domains, one is a catalytic protein tyrosine phosphatase (PTP) domain, where N-terminal SH2 domain binds intramolecularly to PTP site, and the other is a C-terminal tail with two tyrosyl phosphorylation sites [23]. These two SH2 domains regulated catalytic activity and directly block the phosphatase active site. The C-terminal domain is connected to adjacent domains through the peptide backbone. In particular, the N-SH2 domain is a conformational switch, it either binds and inhibits the phosphatase, or it binds phosphoproteins and activates the enzyme, but the C-terminal SH2 domain contributes binding energy and specificity, no direct effect on activation [24].
The development of SHP2 inhibitors has started since 2006 year [25]. Currently, there are several SHP2 inhibitors, including JAB-3068 and TNO155, are under phase I or phase II clinical trials for treating the non-small cell lung cancer and the head and neck cancer in adult patients [26, 27]. Previous studies showed that many SHP2 inhibitors exhibited inhibitory effects in various cancers [28]. Interestingly, the recent preclinical studies reported three SHP2 inhibitors, SHP099, SHP836 and SHP043, which inhibited the lung cancer cells proliferation through suppressing the efficacious related cell signaling pathways in vitro and in vivo. These data about allosteric inhibitors of SHP2 indicate that the inactivation of SHP2's biological function could be a potential therapeutic approach for lung cancer treatment [29-32]. In this review, we summarize the recent preclinical and clinical SHP2 inhibitors studies related to the lung cancer treatment.
2. The preclinical screening of the SHP2 inhibitors 2.1. SHP2/ERK inhibitor: SHP099The extracellular-signal-regulated kinase consists of ERK1 and ERK2 which play critical roles in transmitting the signals from the surface receptors to the nucleus. It has been confirmed that the ERKs also participate in the induction of cellular proliferation and differentiation [33]. Therefore, modulating the signaling pathways that affect ERKs biological function could modulate the target cells' proliferation. Interestingly, previous studies showed the SHP2 mutation affects the RAS/ERK signaling in human cancers including colon cancer, lung cancer and leukemia [34]. The further investigations on the ERK signaling pathway revealed that SHP2 is on the upper stream of ERK. Besides, the ERK phosphorylation is correlated with the gaining of oncogenic functions [35]. SHP2 inhibitor in the colon cancer cell lines or lung cancer cells blocked ERK1/2 phosphorylation and decreased RAS activation [36]. As a cascade reaction, the change of RAS function also triggers the activation of MAPK/RAS pathway [37].
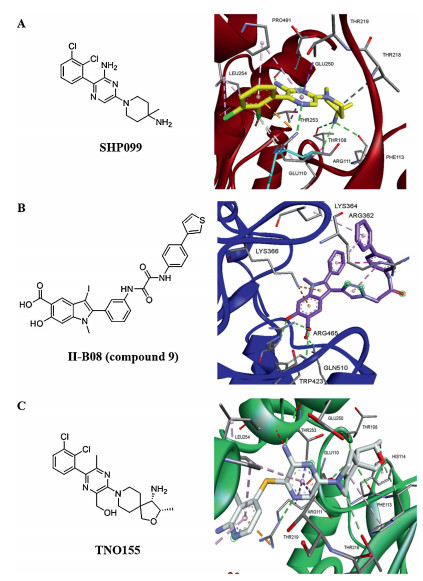
6-(4-Amino-4-methyl-1-piperidinyl)-3-(2, 3-dichlorophenyl)-2-pyrazinamine (SHP099) is a potent SHP2 inhibitor which chemical structure is shown in Fig. 2A. The polar and ionic functional groups in SHP099 are exposed, which increased its selectivity and oral bioavailability significantly [38]. It exhibits inactive SHP2 protein with half maximal inhibitory concentration (IC50) is 0.07 μmol/L and antitumor activity in xenograft models with phospho-ERK (p-ERK) IC50=0.25 μmol/L [30]. Pascal D. Fortin's group obtained the crystal structure of SHP099–SHP2 complex which showed SHP099 bound to the interface of N-terminal, C-terminal and the PTP domains on the SHP2 (Fig. 2A) [31, 39].

|
Download:
|
| Fig. 2. (A) The chemical structure of SHP099 is in the left; Right is X-ray crystallography structure of SHP099 with SHP2 (PDB 5EHR); (B) Left is chemical structure of II-B08; right is co-crystal structure SHP2 with II-B08 (PDB 3O5X); (C) Chemical structure of TNO155 and crystal structure of SHP2 in complex with TNO155 (PDB 7JVM). All crystal structure drawings were made using PyMol (https://pymol.org/2/). | |
PD-1 is frequently up-regulated in tumor cells to inhibit the activity of T-cells and prevent autoimmunity [40]. Studies showed SHP099 magnified the anti-tumor immunity via increasing the proportion of CD8+, interferon gamma (IFN-γ)+ T cells and elevated cytotoxic T-cell related genes. Interestingly, a synergic effect between the SHP099 and anti-PD1 antibody in treating the CT-26 colon cancer has been observed in a mice model [21, 41]. However, there's lack of the report of whether such synergic effect exists in the lung cancer treatment.
2.2. SHP2 E76K inhibitor: #220-324After searching the currently known SHP2 inhibitors in the CADD database, we found the compound #220-324 (Fig. 3) exhibits the highest selectivity among the other SHP2 inhibitors. Specifically, the IC50 value of #220-324 in inhibiting SHP2 is 14 μmol/L. The docking structure of the #220-324/SHP2 complex from Cheng's group revealed that the #220-324 directly binds to the SHP2 catalytic site through hydrophobic contact [42]. Besides, in hematopoietic cells, the #220-324 showed 3 fold more selectivity for SHP2 than SHP1 and TC-PTP which demonstrated that #220-324 is a highly selective SHP2 inhibitor [42].

|
Download:
|
| Fig. 3. Small molecule inhibitors of SHP2 in this review. | |
Previous studies showed that SHP2 E76K mutation has found in lung cancer [43]. To further explore the #220-324's inhibitory effects on SHP2, SHP2 E76K and the mechanisms which conducted in vitro experiments using the purified SHP2 protein tyrosine phosphatase (PTP) domain, wild-type (WT) full-length SHP2 and the full length SHP2 E76K mutant protein. In vitro results showed #220-324 exhibited a dose-dependent inhibitory effect on SHP2 mutant with an IC50 value of 23.81 μmol/L, which is 1.5 fold higher than SHP2 PTP domain. Nevertheless, #220-324 exhibited an inhibitory effect on the SHP2 E76K is 3 fold lower than full-length WT SHP2. In sum, #220-324 exhibits better inhibitory effect on SHP2 E76K than on WT or SHP2 PTP domain.
Besides in vitro protein inhibition, #220-324 showed extraordinary inhibitory effects on the SHP2 E76K juvenile myelomonocytic leukemia (JMML) cells with an IC50 value of 7.24 μmol/L. Furthermore, the IC50 value of #220-324 on the primary cells obtained from the clinical patients (including both WT and SHP2 PTP11 E76K) is 3.09 μmol/L. Strikingly, the exposure of the JMML cells from healthy donors to #220-324 under 100 μmol/L showed no significant inhibition. This result reveals that #220-324 could be a promising targeted agent for lung cancer developing to hematologic malignancy. In addition, SHP2 mutated lung cancer cells were more sensitive to SHP2 inhibitor as well as contrast with WT PTP11 cells [44].
Although the small molecule inhibitor #220-324 showed bioavailability in cancer cells, its affinity to SHP2 is low as the IC50 values obtained in the in vitro experiments above 20 μmol/L. Such a result demonstrates #220-324 is unsuitable for preclinical or clinical trials. More studies are needed for further optimizing of this compound to synthesize better SHP2 inhibitor with sensitive affinity and effective result for solid tumor therapy.
2.3. SHP2/MAPK inhibitor: compound 23Allosteric SHP2 inhibitor compound 23, chemical name is 1-(4-(6-bromonaphthalen-2-yl) thiazol-2-yl)-4-methylpiperidin-4-amine (Fig. 3). It is similar to SHP099, binds to the closed conformation of SHP2 on the locks interface of the N-terminal, C-terminal SHP2 and phosphatase domain [45].
The synthesis of compound 23 based on the structure of compound 9 (II-B08, Fig. 2B). Because the hydrogen bonds are vital for the interaction, the phenyl of the compound 9 was changed with several common aryl groups, particularly replaced with biphenyl or naphthyl form. The compound 23 exhibited inhibitory effects on SHP2 E76A with an IC50 value of 0.7 μmol/L. But compound 23 showed no inhibitory effect on SHP2 PTP under in vitro condition. The structure–activity relationship study on selectivity profiling identifies that compound 23 exhibited a 48 fold higher selectivity for SHP2 over the homologous SHP2 and exhibited more than 30 fold preference for the SHP2 PTP over other PTPs [46].
Studies showed compound 23 exposures in the large cell lung cancer cell (NCI-H661) of SHP2 N58S mutation with an IC50 value of 1.2 μmol/L. Besides, the downregulation of the phosphorylation of ERK/AKT pathway has also been observed in the same cell line. In the H1975 human NSCLC cell line with SHP2 T90M mutation, compound 23 exposure diminished the RAS expression, which demonstrated that compound 23 suppressed the MAPK signaling pathway. Moreover, compound 23 treated SF268 cell line (neuroblastoma cell line) showed that SHP2 is physically associated with the oncoprotein YAP and the tyrosine phosphorylation of YAP is regulated by the catalytic function of SHP2 PTP. Indeed, compound 23 effects on A549 cells (adenocarcinomic human alveolar basal epithelial cells) with KRAS mutation showed YAP phosphorylation is stimulated [47].
The pharmacokinetics results showed that compound 23has good oral absorption, [10 mg/kg, po area under the plasma concentration time curve (AUC): 9860 nmolh/L) and 67% F bioavailability with a half-life of 13.3 h. Subcutaneous transplantation of MV4-11 cells into immunodeficient mice established a xenograft mice model that treated with 10 or 30 mg/kg of compound 23 one day could induce tumor growth, and tumor volume was decreased. This result of the mice model explained compound 23 served well anti-tumor effect in vivo [47].
In sum, the current studies demonstrated that compound 23 inhibits various lung cancer cell lines including large lung cell carcinoma cancer cell line and drug-resistant NSCLC cell lines. Further studies revealed that compound 23 blocks RAS/MAPK and ERK/AKT activation in the cancer cells. However, compound 23 is unstable in vivo. Further investigations on compound 23 are needed to better understand its effects under in vivo conditions for further optimization.
2.4. SHP2/GRB2-associated-binding protein 1 (Gab-1) inhibitor: fumosorinoneMost SHP2 inhibitors are gained from selective chemical synthesis, while a few SHP2 inhibitors have been extracted from microorganisms, especially from insect pathogenic fungi. Therefore, insect pathogenic fungi have been served as emerging resources of small molecule PTP inhibitors. The compound fumosorinone (Fumos) is identified from an insect pathogenic fungi in a recent study [48]. Studies showed that Fumos inhibited the expression of PTP1B, increased glucose uptake, stimulated insulin signaling pathway and induced insulin resistance in type 2 diabetes [49]. Luo's group study showed Fumos displayed the high selective inhibition on SHP2 over other human PTPs [25]. The chemical structure of Fumos is shown in Fig. 3.
Fumos is essentially a pyrrolidine alkaloid (Fig. 3), which inactivates the PTP activity of SHP2 with an IC50 value of 6.31 μmol/L under in vitro conditions. Interestingly, the IC50 value of Fumos on the full-length WT SHP2 is 26.52 μmol/L, which demonstrates the PTP domains of SHP2 are the target site of Fumos. The computer docking and structural analysis of Fumos–SHP2 complex showed that there are four hydrogens formed via Fumos binds with Gly464, Arg465, Ala461 and Ser460 on the PTP domain of SHP2, as well as two hydrophobic joints to Cys459 and Arg465 in the PTP loop. Besides, Fumos also enable to form a hydrogen bond with Gly427 in the WPD loop and with Tyr279 in phosphotyrosine in the recognition loop. These results of computer docking suggest that Fumos is an SHP2 specific inhibitor. Combined with the analysis of Line weaver-Burk result illustrates Fumos decreased the maximum reaction rate (Vmax) without affecting the Michaelis constant (Km). All the above results demonstrate Fumos is a noncompetitive SHP2 inhibitor that follows the Michaelis-Menten equation [25].
In HeLa and HEK293T cell lines, the ERK1/2 phosphorylation is significantly decreased in the presence of Fumos with epidermal growth factor (EGF) simulation. Similarly, the study about SHP2 and RAS showed SHP2 plays a role upstream of RAS, the PTP activity of SHP2 required for RAS fully activated. Fumos inhibits RAS activation both in HeLa and HEK293T cells line [25]. Base on the results mentioned above, we came with the conclusion that SHP2 functions in the upstream of RAS in the RAS/MAPK kinase signaling pathway.
The Gab family of docking proteins (Gab1 and Gab2) is phosphorylated in response to the binding of various cytokines and growth factors to the receptors on the cell surface. Gab1 mediates the generation of branched tubules and plays a key role in cell growth response, transformation and apoptosis. Gab2 is scaffolding protein which acts downstream of several membrane receptors including cytokine, antigen, hormone, cell matrix and growth factor receptors to regulate multiple signaling pathways [50, 51]. Gab1 or Gab2 binds to SHP2 activates RAS/ERK in EGF-stimulated cells [52]. The Luo's group's study showed the treatment of Fumos in HEK293T and MDA-MB-231 increased the tyrosine-phosphorylation of Gab1 and Fumos inhibited the invasion of tumor cells with a dose-dependent manner in HeLa and MDA-MB-231 cells. Surprisingly, no obvious toxicity effects were observed in the experiment. It is worth mentioning that because Fumos does not affect ERK1/2 [25]. It can be used as an alternative option for SHP2 related lung cancer patients.
2.5. SHP2/RAS inhibitor: RMC-4550The chemical structure of inhibitor RMC-4550 is shown in Fig. 3. Focused on multiparameter optimization using SBDD for enhancing physicochemical properties, cell potency, selectivity and rodent oral bioavailability, RMC-4550 was synthesized in 5 linear (6 total) steps from the readily accessible or commercially available intermediates [53]. Now it is seen as another potent, selective and orally bioavailable allosteric inhibitor.
The IC50 of RMC-4550 on full-length SHP2 enzyme is 1.55 nmol/L with no inhibition on the catalytic domain. Such result indicates the RMC-4550 is highly selective for full-length SHP2 and exhibits high quality, drug-like preclinical profile. Studies showed that the RMC-4550 inhibited Calu-1 (IC50=7 nmol/L) and PC9 (IC50=39 nmol/L). Further mechanism studies revealed that RMC-4550 decreased the ERK phosphorylation in the two cell lines. Strikingly, RMC-4550 inhibited NCI-H358 (KRAS G12C/+) and MIA PaCa-2 (pancreas, KRAS G12C/G12C) through down regulating the RAS–guanosine triphosphate (GTP) and p-ERK levels in the two cell lines. Studies also showed the RMC-4550 activates caspase 3/7 in NCl-H358 cells (NSCL cell line) [54, 55].
RMC-4550 inhibits NCl-H1838 proliferation through downregulating the RAS-GTP level and suppressing ERK phosphorylation. In the EGFR-driven KYSE-520 human esophageal cancer xenograft model, RMC-4550 displayed a dose-dependent efficacy which is consistent with target modulation, assessed by p-ERK inhibition in tumors. RMC-4550 is well tolerated at doses that achieved maximal and sustained efficacy in KYSE-520 human esophageal cancer xenograft model. In summary, RMC-4550 exemplifies a novel class of potent allosteric inhibitors of SHP2 with an excellent drug-like property profile [55].
2.6. SHP2/ERK1/2/AKT inhibitors: II-B08 and 11a-1II-B08 (compound 9) inhibitor (Fig. 2B) is constructed from Salicylic Acid-based Combinatorial library. The key synthesized step is the salicylic acid core 1 formed by eight steps with Sonogashira coupling and an electrophilic cyclization. As shown in Scheme 1, precursor compound 2 was the esterification of 4-aminosalicylic acid with dimethyl sulfate product after isolated in 86% yield, compound 3 was made from compound 2 combined with paraformaldehyde and NaCNBH3 and with iodine gave methyl 4-(dimethylamino)-2-hydroxy-5-iodobenzoate 4 to form compound 5, then compound 6 was synthesized by electrophilic cyclization of compound 5. The major compound 7 was exposed through compound 6 with trimethylsilyacetylene which is another Pd(0)-catalyzed Sonogahira coupling. At last, TBAF and KOH were used to remove the protective groups in compound 7 sequentially to afford the salicylic acid core 1 [56]. The crystal structure of SHP2 in complex with II-B08 showed the inhibitor binds to SHP2 active-site pocket and forms extensive interactions with residues in the phosphor-tyrosine (pTyr) recognition loop, P-loop and WPD loop, the phenolic oxygenatom O1 makes a hydrogen bond with the amide R465 in the P-loop (Fig. 2A) [57-60].

|
Download:
|
| Scheme 1. Synthesis of compound 9. | |
II-B08 inactivates SHP2 with an IC50 value of 5.5 μmol/L. The kinetic analysis indicated that II-B08 is a reversible and non-competitive inhibitor for SHP2 in vitro. The crystallographic analysis of II-B08/SHP2 complex reveals that the salicylic acid core occupies the PTP domain (a salicylic acid-based small molecule inhibitor for SHP2). In HEK293T cells, II-B08 strongly inhibits the sustained phase of ERK1/2 activation after EGF stimulation. The same effect was observed in the NIH3T3 cell. What is more, the studies using the hematopoietic progenitors from JMML patients indicated that II-B08 decreased the granulocyte-macrophage colony-stimulating factor (GM-CSF) stimulated hypersensitivity induced by SHP2 mutants, especially in SHP2/E76K and SHP2/D61Y hematopoietic progenitors.
A structure-guided and fragment-based library was used to obtained hydroxyindole carboxylic acid-based SHP2 inhibitor 11a-1. The II-B08 and 11a-1 structures were used as references to guide the synthesis of 11a-1 (Fig. 3). The compound 11a-1 exhibited the lowest IC50 value of 0.2 μmol/L against SHP2. What is more, studies showed the 11a-1 exhibited over 5 fold selectivity for SHP2 compare with a large panel of other mammalian PTPs, including protein-tyrosine phosphatase 1B (PTP1B), T-cell protein tyrosine phosphatase (PTPN2) and PTPH1 (PTPN3). As we mentioned earlier, II-B08 binds to SHP2 active site, while 11a-1 binds deeply into the active site with recognition of pTyr cleft, occupies largely active pocket and adjacent α-phenyl ring forms strong π-π tacking with Y279 in the pTyr recognition loop [59, 61].
The inhibition activity of 11a-1 was confirmed in several cancer cell lines including NSCLC cell H1975 and breast cancer cell SKBR3. The 11a-1 inhibited H1975 proliferation with a dose-dependent manner (IC50=0.17 μmol/L). Further studies revealed the 11a-1 inactivated the SHP2 function, therefore significantly reduced the ERK1/2. Meanwhile, 11a-1 upregulated the paxillin (Py118) phosphorylation. The same results were observed in the SKBR3 cell. Besides, 11a-1 was found more potent in inhibiting oncogenic KITD814 bearing cells compared to II-B08 in various cancers including gastrointestinal stromal tumors, acute myelogenous leukemia and systemic mastocytosis. What is more, 11a-1 suppresses the activation of AKT and ERK1/2 in melanoma and non-small cell lung cancer cell lines. Collectively, all the 11a-1 studies demonstrated that it is highly potent in inhibiting the proliferation of H1975, SKBR3 and oncogenic KITD814V bearing myeloid cells via deactivating SHP2 function. The 11a-1 has great potential to be applied in the clinic to treat non-small cell lung cancer or pulmonary metastatic cancer in future [61-64].
2.7. SHP2 inhibitor: cryptotanshinoneCryptotanshinone was identified through computer-aided drug design (CADD) screening using SHP2 enzymatic assays. Besides, the cryptotanshinone has been applied in treating the cardiovascular diseases in Asian countries. It is one of the components of a traditional medicinal herbal plant Salvia miltiorrhiza Bunge (Dan shen) (Fig. 3) [27, 65]. Previous studies showed the cryptotanshinone inhibited the SHP2 catalyzed hydrolysis (PTP domain) under in vitro condition with the IC50 value of 22.5 μmol/L. Such a value is 1.76 fold more sensitive for SHP2 over its homologous proteins SHP1 and PTP1B. Also, the cryptotanshinone suppressed the full-length WT SHP2 with IC50 value of 45.18 μmol/L and the SHP2 E76K mutant with IC50 value of 23.9 μmol/L. The above results plus the computer docking result indicated that the cryptotanshinone binds to the two carbonyl oxygen atoms with Lys364 and Lys366 at the SHP2 catalytic site. These results demonstrated that the cryptotanshinone binds to the N-SHP2 forming a closed conformation which deactivates SHP2 activity [66].
Previous studies illustrated that the SHP2 induces IL3 stimulated cell proliferation through activating ERK, AKT and JAK2 which is confirmed by the finding that the bone marrow cells from JMML patients or mouse with SHP2 mutation are sensitive to IL-3 and GM-CSF. Therefore, cryptotanshinone exhibited significant inhibitory effect in Ba/F3 cells with an IC50 value of 17.22 μmol/L. The cryptotanshinone exposure significantly decreases the phosphorylation levels of ERK, AKT, JAK2 and STAT5. The IL-3 treatment also significantly decrease the tyrosine phosphorylation of Gab2 in the Ba/F3 cells (pro-B cell line). The cryptotanshinone significantly inhibited the proliferation of HeLa cells with dose dependence. Cryptotanshinone also showed inhibitory effects in PTPN11E76K/+ mutant mouse embryonic fibroblasts (MEKs), human lymphoma cell U937 harboring SHP2G60R mutant and H661 with SHP2N58S mutant via a dose-dependent manner [66, 67].
In summary, the cryptotanshinone inhibits SHP2 catalytic activity through downregulating RAS/ERK, PI3K/AKT and JAK2/STAT5 pathways. Since the SHP2 depleted cells presented much less sensitivity to cryptotanshinone indicates that cryptotanshinone may as alternative targets. A robust apoptosis-inducing effect on cryptotanshinone treated U937 cells and G1-S arrest on H661 cells suggest that cryptotanshinone treatment may be a potential alternative therapy for the PTPN11 mutant patients suffering from lung cancer with bone marrow metastasis [68, 69].
3. Patent inhibitor 3.1. NSC-87877 and NSC-1171998-Hydroxy-7-(6-sulfonaphthalen-2-yl)-diazenyl-quinoline-5-sulfonic acid (NSC-87877) was constructed through virtual screening of the NCl Diversity Set chemical library combine with experiments (Fig. 3). The continuous optimization and restructuring ensured the NSC-87877, which contains naphthalene-sulfonic acid and quinolinesulfonic acid as a potent inhibitor of SHP2 target with an IC50 value of 0.318 μmol/L. It showed approximately 5 and 24-fold selectivity for SHP2 over its homologous proteins PTP1B and HePTP respectively. The docking result suggested that NSC-87877 bound to the catalytic domain of SHP2 and the a ring sulfonic acid conjoin the side chain NH3 group of Lys280 and the side chain NH2 group of Asn281 through hydrogen bonds, the B ring sulfonic acid group bound with the backbone NH group of Arg465 [70-72].
Studies showed the NSC-87877 reduced the basal SHP2 PTP activity in the HEK293 cell with EGF stimulation. Besides, the further studies revealed that the NSC-87877 decreased EGF induced ERK1/2 activation in a dose-dependent manner and it also inhibits ERK 1/2 activation through forming an NSC-87877–Gab1–SHP2 complex. However, HEK293 cells treated with NSC-87877 associated Gab1–SHP2 showed no inhibition on the Gab1 tyrosine phosphorylation. A possible explanation is NSC-87877 off-targets the SHP2 activation pathway. Interestingly, NSC-87877 inhibits the EGF stimulated SHP2 PTP, RAS and ERK1/2 activation in the human breast carcinoma cell MDA-MB-468 expressing SHP2 and SHP1.The time-course experiments results showed no inhibitory effects on the tested cells until 2–3 h post adding NSC-87877, indicating it might went through an activation process. Further investigation is needed to better understand the mechanisms of NSC-87877 in cancer cell lines [72, 73].
The NSC-117199 chemical name is 3-[2-(2-nitrophenyl) hydrazinyl]-2-oxoindole-5-sulfonic acid. It binds to the catalytic site of SHP2 same as NSC-87877 binds. The NSC-117199 structure (with the isatin core) was shown in Fig. 3. The other role of NSC-117199 is similar to NSC-87877 [74-77].
3.2. The SHP2 analogs: SPI-112 and SPI-112MeNSC-11799 was a leader compound to synthesis SPI-112 form SHP2 inhibitor. Its chemical name is (Z)-3-(2-(5-(N-(4-fluorobenzyl)sulfamoyl)-2-oxoindolin-3-ylidene) hydrazinyl) benzoic acid (Fig. 3). To improve the poor cell-permeability of NSC-11799, a methyl ester bound to SPI-112 composed SPI-112Me (Fig. 3). It can be hydrolyzed to SPI-112 upon entry into cells. The SPI effectively inhibited SHP2 PTP with an IC50 value of 1.0 μmol/L, methyl ester prodrug SPI-112Me inhibit SHP2 PTPs. The computer docking analysis indicated that the SPI-112 binds to the Gly464 and Arg465 of the P-loop in the catalytic pocket of SPH2 through hydrogen bonds. Further studies showed that the SPI-112Me inhibits ERK1/2 activation through forming a Gab1-SHP2 chimaera [71, 78].
SHP2 is activated in MDA-MB-468 breast cancer cells via EGF stimulation. The SPI-112Me preincubation reduced the EGF stimulated SHP2 PTP activity in MDA-MB-468 cell. However, no effects were observed when treat with SPI-112 in the same way. Seeing the study showed cytokine-dependent TF-1 myeloid cells into cytokine independence by upregulate B-cell lymphoma extra-large (Bcl-xL) through the ERK1/2 pathway, SPI-112Me decreased the expression of p-ERK1/2 and Bcl-xL in TF-1/SHP2 E76K cells with dose dependent. Furthermore, SPI-112Me enhances the IFN-γ induced STAT1 tyrosine phosphorylation in HT-29 cells. Besides, IFN-γ and SPI-112 showed a synergic effect in inhibiting HT29 cells proliferation [71, 79].
4. Clinical trial 4.1. SHP2 inhibitor: TNO155The TNO155 inhibits SHP2 with an IC50 value of 0.011 μmol/L, which is 100 fold greater potency than SHP099 in vitro. It inhibits the SHP2 mediated MAPK signaling pathway and the cancer cells expressing SHP2 [80]. The César Serrano García group's results showed that the combination of TNO155 and PD1 activates the RAS–RAF–ERK signaling pathway showed excellent results. The TNO155 oral bioavailability in mouse and rat is 78% and 86%, respectively [81].
The crystal structure of TNO155 with SHP2 revealed extended amine directly jointed with residues E110, S109 and F113. The pyrazineaniline interacts with E250 in a hydrogen bond, while R111 participates in the superposition interaction with the cation pi of the chloropyridine ring, and pre-organized by hydrogen bonding through pyrazine N [82]. The co-crystal structure is shown in Fig. 2C.
TNO155 is currently in phase I clinical trials and is tested alone or combination with EGF816 (nazartinib) in advanced EGFR mutant NSCLC, KRAS G12-mutant NSCLC, esophageal squamous cell cancer (SCC), head/neck SCC and melanoma patients. However, in the phase I/II trials of MRTX849 (KRAS G12C inhibitor) combined with TNO155 in patients with advanced solid tumors and KRAS G12C mutation is still recruiting volunteers. On the other hand, the phase Ib study of TNO155 in combination with spartalizumab or ribociclib in the selected malignancies will be administered until the subjects' experiences unacceptable toxicity, progressive disease, and/or has treatment discontinued at the discretion of the investigator or the subject, or due to the withdrawal of consent [83-85].
4.2. SHP2 inhibitor: JAB-3068JAB-3068 is a small molecule inhibitor of SHP2 developed by Jacobio Pharmaceuticals for NSCLC, head and neck cancer, esophageal cancer and other metastatic solid tumors. It has entered into phase II 1/2a clinical trials and recruited 120 participants of age 18 years old or older with advanced solid tumors. Phase I clinical trials were conducted in the United States but phase II clinical trials are all in China. Specifically, the JAB-3068 is administered to the patients orally in the morning, following a fast of approximately 6 h before on pharmacokinetics collection. Patients will continue to fast for approximately 2 h after the administration of JAB-3068. On non-PK days patients will fast about 2 h before JAB-3068 and continue to fast for approximately 2 h afterwards. This phase of clinical research is expected to be completed in September 2020 [28, 86, 87].
JAB-3068 binds to and suppresses SHP2, therefore interferes the MAPK signaling pathway and inhibits the proliferation of the tumor cells expressing SHP2. Since SHP2 also regulates PD-1 mediated signal transduction and anti-PD-1 has a significant effect, the recognition of the role of SHP2 inhibitors-JAB-3068 combined with immune checkpoint inhibitors in cancer immunotherapy may improve the treatment of cancer patients [88, 89].
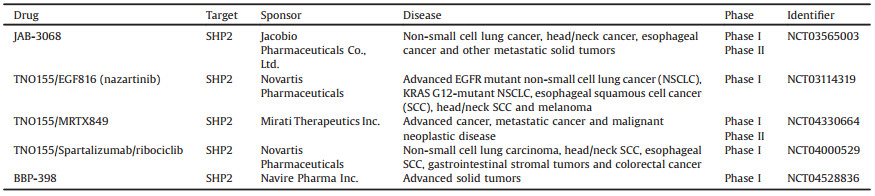
4.3. SHP2 inhibitor: BBP-398BBP-398 formerly known as IACS-15509, it is currently the latest SHP2 inhibitor and has entered clinical trials in phase I stage. The subjects are mainly from advanced or metastatic KRASG12C mutant non-small cell lung patients, KRASG12C mutant solid tumor other than NSCLC, solid tumor with other MAPK pathway alterations (excluding BRAF V600X) and EGFR-mutant NSCLC [90]. Unfortunately, like JAB-3068, its chemical structure has not yet been fully announced, their structures will be made public soon. The emergence of these clinical trial drugs provides more options for the treatment of SHP2 related diseases (Table 1).
|
|
Table 1 Clinical trials SHP2 inhibitors used in this review. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The recent progresses of SHP2 inhibitor studies are exciting. Most studies indicated that the lower concentration and effective treatment of lung cancer cells with minimal cytotoxicity to improve the remission and survival rate of patients is urgently needed. The inhibitor #220-324, cryptotanshinone, II-B08 or SPI are not optimal drugs for clinical application because their high IC50 values indicate their high cytotoxicity. Small molecule inhibitor SHP099 and RMC-4550 are expected to become candidate drugs but the in vivo studies are needed to understand their mechanisms. Thus, following the clinical trials, targeting the SHP2 alone or in combination with immunosuppression or other chemotherapy drugs is a new strategy. In patients with NSCLC or other subtypes of lung cancer, the dose-expansion of JAB-3068 study is designed to evaluate its antitumor activity is going through clinical Phase II trials.
At present, most SHP2 inhibitors are in the preclinical study stage. Further studies on these SHP2 inhibitors are required to reduce their cytotoxicity and improve the SHP2 selectivity. The SHP2 dual-target drugs have potential to be developed as an alternative option of lung cancer treatment. The inhibition of SHP2-KRAS targets might have curative effects in the treatment of double-mutated lung cancer. Furthermore, various drug design methods were used in SHP2 drug discovery, as exemplified fragment based drug design was applied in II-B08, RMC-4550 and #220-324 were synthesized by SBDD and CADD screening. Significantly, the application of these innovative solutions, such as quantum chemistry, and virtual target profiling could accelerate drug development; meanwhile the new techniques photodynamic immunotherapy combined with small molecule inhibitors is a trend in the treatment of lung cancer and other malignant tumors [91, 92].
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 81904178, 82073311) and "Hundred Talents Program" of the Hospital of Chengdu University of Traditional Chinese Medicine (Nos. 20-Q03, 20-Q08).
| [1] |
C. Zappa, S.A. Mousa, Transl. Lung Cancer Res. 5 (2016) 288-300. DOI:10.21037/tlcr.2016.06.07 |
| [2] |
N. Yasui, G.M. Findlay, G.D. Gish, et al., Mol. Cell 54 (2014) 1034-1041. DOI:10.1016/j.molcel.2014.05.002 |
| [3] |
M. Kim, M. Baek, D.J. Kim, Curr. Pharm. Des. 23 (2017) 4226-4246. |
| [4] |
V.E. Schneeberger, N. Luetteke, Y. Ren, et al., Carcinogenesis 35 (2014) 1717-1725. DOI:10.1093/carcin/bgu025 |
| [5] |
L. He, Y. Li, X. Huang, H. Cheng, Y. Ke, et al., OncoTargets Ther. 12 (2019) 5897-5906. DOI:10.2147/OTT.S210223 |
| [6] |
L. Jiang, W. Xu, Y. Chen, Y. Zhang, Artif. Cells Nanomed. Biotechnol. 47 (2019) 3231-3238. DOI:10.1080/21691401.2019.1646748 |
| [7] |
S. Mainardi, A. Mulero-Sánchez, A. Prahallad, et al., Nat. Med. 7 (2018) 961-967. |
| [8] |
A. Saiardi, R. Bhandari, A.C. Resnick, A.M. Snowman, S.H. Snyder, Science 5704 (2004) 2101-2105. |
| [9] |
Y. Shi, Cell 139 (2009) 468-484. DOI:10.1016/j.cell.2009.10.006 |
| [10] |
A. Alonso, J. Sasin, N. Bottini, et al., Cell 117 (2004) 699-711. DOI:10.1016/j.cell.2004.05.018 |
| [11] |
E.N. Gurzov, W.J. Stanley, T.C. Brodnicki, H.E. Thomas, Trends Endocrinol. Metab. 26 (2015) 30-39. DOI:10.1016/j.tem.2014.10.004 |
| [12] |
L.I. Pao, K. Badour, K.A. Siminovitch, B.G. Neel, Annu. Rev. Immunol. 25 (2007) 473-523. DOI:10.1146/annurev.immunol.23.021704.115647 |
| [13] |
R.H. Reddy, H. Kim, S. Cha, B. Lee, Y.J. Kim, J. Microbiol. Biotechnol. 27 (2017) 878-895. DOI:10.4014/jmb.1701.01079 |
| [14] |
L. Li, X. Xu, Y. Du, et al., Toxicol. Appl. Pharmacol. 399 (2020) 115053. DOI:10.1016/j.taap.2020.115053 |
| [15] |
Y.M. Agazie, M.J. Hayman, Mol. Cell. Biol. 23 (2003) 7875-7886. DOI:10.1128/MCB.23.21.7875-7886.2003 |
| [16] |
J. Zhang, F. Zhang, R. Niu, J. Cell. Mol. Med. 19 (2015) 2075-2083. DOI:10.1111/jcmm.12618 |
| [17] |
T.J. Bauler, N. Kamiya, P.E. Lapinski, et al., Dis. Model. Mech. 4 (2011) 228-239. DOI:10.1242/dmm.006130 |
| [18] |
M. You, D.H. Yu, G.S. Feng, Mol. Cell. Biol. 19 (1999) 2416-2424. DOI:10.1128/MCB.19.3.2416 |
| [19] |
Z. Liu, Y. Zhao, J. Fang, et al., Oncotarget 8 (2017) 53518-53530. DOI:10.18632/oncotarget.18591 |
| [20] |
Y.J. Sun, Z.L. Zhuo, H.P. Xian, et al., Oncotarget 8 (2017) 91123-91133. DOI:10.18632/oncotarget.20249 |
| [21] |
M. Zhao, W. Guo, Y. Wu, et al., Acta Pharm. Sin. B 9 (2019) 304-315. DOI:10.1016/j.apsb.2018.08.009 |
| [22] |
Z.Y. Zhang, Curr. Opin. Chem. Biol. 5 (2001) 416-423. DOI:10.1016/S1367-5931(00)00223-4 |
| [23] |
P. Hof, S. Pluskey, S. Dhe-Paganon, M.J. Eck, S.E. Shoelson, Cell 92 (1998) 441-450. DOI:10.1016/S0092-8674(00)80938-1 |
| [24] |
E. Darian, O. Guvench, B. Yu, et al., Proteins 79 (2011) 1573-1588. DOI:10.1002/prot.22984 |
| [25] |
L. Chen, S.S. Sung, M.L. Yip, et al., Mol. Pharmacol. 70 (2006) 562-570. DOI:10.1124/mol.106.025536 |
| [26] |
R.A. Cerulli, J.A. Kritzer, Org. Biomol. Chem. 18 (2020) 583-605. DOI:10.1039/C9OB01998G |
| [27] |
F.I. Nollmann, D.A. Ruess, Biomedicines 8 (2020) 281. DOI:10.3390/biomedicines8080281 |
| [28] |
X. Yuan, H. Bu, J. Zhou, C.Y. Yang, H. Zhang, J. Med. Chem. 63 (2020) 11368-11396. DOI:10.1021/acs.jmedchem.0c00249 |
| [29] |
T.A. Ahmed, C. Adamopoulos, Z. Karoulia, et al., Cell Rep. 26 (2019) 65-78. DOI:10.1016/j.celrep.2018.12.013 |
| [30] |
J.G. Fortanet, C.H. Chen, Y.N. Chen, et al., J. Med. Chem. 59 (2016) 7773-7782. DOI:10.1021/acs.jmedchem.6b00680 |
| [31] |
Y.N. Chen, M.J. LaMarche, H.M. Chan, et al., Nature 535 (2016) 148-152. DOI:10.1038/nature18621 |
| [32] |
S. De Munter, M. Köhn, M. Bollen, ACS Chem. Biol. 8 (2013) 36-45. DOI:10.1021/cb300597g |
| [33] |
M. Cargnello, P.P. Roux, Microbiol. Mol. Biol. Rev. 75 (2011) 50-83. DOI:10.1128/MMBR.00031-10 |
| [34] |
X. Chen, X. Fu, W. Zhao, et al., Signal Transduct. Target. Ther. 5 (2020) 83. DOI:10.1038/s41392-020-0192-0 |
| [35] |
Z.Q. Shi, D.H. Yu, M. Park, M. Marshall, G.S. Feng, Mol. Cell. Biol. 20 (2000) 1526-1536. DOI:10.1128/MCB.20.5.1526-1536.2000 |
| [36] |
H. Lu, C. Liu, R. Velazquez, et al., Mol. Cancer Ther. 18 (2019) 1323-1334. DOI:10.1158/1535-7163.MCT-18-0852 |
| [37] |
L. Santarpia, S.M. Lippman, A.K. El-Naggar, Expert Opin. Ther. Targets 16 (2012) 103-119. DOI:10.1517/14728222.2011.645805 |
| [38] |
P. Sarver, M. Acker, J.T. Bagdanoff, et al., J. Med. Chem. 62 (2019) 1793-1802. DOI:10.1021/acs.jmedchem.8b01726 |
| [39] |
J.R. LaRochelle, M. Fodor, V. Vemulapalli, et al., Nat. Commun. 9 (2018) 4508. DOI:10.1038/s41467-018-06823-9 |
| [40] |
L.M. Francisco, P.T. Sage, A.H. Sharpe, Immunol. Rev. 236 (2010) 219-242. DOI:10.1111/j.1600-065X.2010.00923.x |
| [41] |
W.J. Guo, Q. Xu, J. Pharmacol. Sci. 144 (2020) 139-146. DOI:10.1016/j.jphs.2020.06.002 |
| [42] |
B. Yu, W. Liu, W.M. Yu, et al., Mol. Cancer Ther. 12 (2013) 1738-1748. DOI:10.1158/1535-7163.MCT-13-0049-T |
| [43] |
X. Liu, H. Zheng, X. Li, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 984-989. DOI:10.1073/pnas.1508535113 |
| [44] |
H. Chen, S. Libring, K.V. Ruddraraju, et al., Oncogene 39 (2020) 7166-7180. DOI:10.1038/s41388-020-01488-5 |
| [45] |
P. Parsonidis, M. Shaik, A.P. Serafeim, et al., Molecules 24 (2019) 4389. DOI:10.3390/molecules24234389 |
| [46] |
W.S. Liu, W.Y. Jin, L. Zhou, et al., J. Comput. Aided. Mol. Des. 33 (2019) 759-774. DOI:10.1007/s10822-019-00213-z |
| [47] |
J. Xie, X. Si, S. Gu, et al., J. Med. Chem. 60 (2017) 10205-10219. DOI:10.1021/acs.jmedchem.7b01520 |
| [48] |
C. Chen, T. Xue, P. Fan, et al., Oncol. Lett. 15 (2018) 10055-10062. |
| [49] |
Z.Q. Liu, T. Liu, C. Chen, et al., Toxicol. Appl. Pharmacol. 285 (2015) 61-70. DOI:10.1016/j.taap.2015.03.011 |
| [50] |
B. Lamothe, M. Yamada, U. Schaeper, et al., Mol. Cell. Biol. 24 (2004) 5657-5666. DOI:10.1128/MCB.24.13.5657-5666.2004 |
| [51] |
G.A. Rodrigues, M. Falasca, Z. Zhang, S.H. Ong, J. Schlessinger, Mol. Cell. Biol. 20 (2000) 1448-1459. DOI:10.1128/MCB.20.4.1448-1459.2000 |
| [52] |
Y. Ren, S. Meng, L. Mei, et al., J. Biol. Chem. 279 (2004) 8497-8505. DOI:10.1074/jbc.M312575200 |
| [53] |
R.R. Wang, W.S. Liu, L. Zhou, Y. Ma, R.L. Wang, J. Biomol. Struct. Dyn. 38 (2020) 1525-1538. DOI:10.1080/07391102.2019.1613266 |
| [54] |
R.J. Nichols, F. Haderk, C. Stahlhut, et al., Nat. Cell. Biol. 20 (2018) 1064-1073. DOI:10.1038/s41556-018-0169-1 |
| [55] |
J. Gillson, Y. Ramaswamy, G. Singh, et al., Cancers (Basel) 12 (2020) 1341. DOI:10.3390/cancers12051341 |
| [56] |
X. Zhang, Y. He, S. Liu, et al., J. Med. Chem. 53 (2010) 2482-2493. DOI:10.1021/jm901645u |
| [57] |
A.J. Barr, Future Med. Chem. 2 (2010) 1563-1576. DOI:10.4155/fmc.10.241 |
| [58] |
S. Butterworth, M. Overduin, A.J. Barr, Future Med. Chem. 6 (2014) 1423-1437. DOI:10.4155/fmc.14.88 |
| [59] |
L.F. Zeng, R.Y. Zhang, Z.H. Yu, et al., J. Med. Chem. 57 (2014) 6594-6609. DOI:10.1021/jm5006176 |
| [60] |
Q. Wang, W.C. Zhao, X.Q. Fu, Q.C. Zheng, Front. Chemistry 8 (2020) 1059. |
| [61] |
R.Y. Zhang, Z.H. Yu, L. Zeng, et al., Oncotarget 7 (2016) 73817-73829. DOI:10.18632/oncotarget.12074 |
| [62] |
R. Tsutsumi, H. Ran, B.G. Neel, FEBS Open Bio. 8 (2018) 1405-1411. DOI:10.1002/2211-5463.12493 |
| [63] |
Z.H. Yu, R.Y. Zhang, C.D. Walls, et al., Biochemistry 53 (2014) 4136-4151. DOI:10.1021/bi5002695 |
| [64] |
Y. Zhao, X. Zhang, K. Guda, et al., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 2592-2597. DOI:10.1073/pnas.0914884107 |
| [65] |
H. Luo, C.T. Vong, H. Chen, et al., Chin. Med. 14 (2019) 48. |
| [66] |
W. Liu, B. Yu, G. Xu, et al., J. Med. Chem. 56 (2013) 7212-7721. DOI:10.1021/jm400474r |
| [67] |
R.J. Chan, M.B. Leedy, V. Munugalavadla, et al., Blood 105 (2005) 3737-3742. DOI:10.1182/blood-2004-10-4002 |
| [68] |
S. Liu, Z. Han, A.L. Trivett, et al., Cancer Immunol. Immunother. 68 (2019) 1059-1071. DOI:10.1007/s00262-019-02326-8 |
| [69] |
L. Jin, Z. Wu, Y. Wang, X. Zhao, Chin. Med. 15 (2020) 66. DOI:10.1186/s13020-020-00348-4 |
| [70] |
C.G. Vazhappilly, E. Saleh, W. Ramadan, et al., Invest. New Drugs 37 (2019) 252-261. DOI:10.1007/s10637-018-0626-5 |
| [71] |
L. Chen, D. Pernazza, L.M. Scott, et al., Biochem. Pharmacol. 80 (2010) 801-810. DOI:10.1016/j.bcp.2010.05.019 |
| [72] |
L.M. Scott, H.R. Lawrence, S.M. Sebti, N.J. Lawrence, J. Wu, Curr. Pharm. Des. 16 (2010) 1843-1862. DOI:10.2174/138161210791209027 |
| [73] |
N. Hanna, A. Montagner, W.H. Lee, et al., FEBS Lett. 580 (2006) 2477-2482. DOI:10.1016/j.febslet.2006.03.088 |
| [74] |
C. Chen, M. Cao, S. Zhu, et al., Sci. Rep. 5 (2015) 17626. DOI:10.1038/srep17626 |
| [75] |
K. Hellmuth, S. Grosskopf, C.T. Lum, et al., Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 7275-7280. DOI:10.1073/pnas.0710468105 |
| [76] |
H.R. Lawrence, R. Pireddu, L. Chen, et al., J. Med. Chem. 51 (2008) 4948-4956. DOI:10.1021/jm8002526 |
| [77] |
C.K. Qu, Cell Res. 10 (2000) 279-288. DOI:10.1038/sj.cr.7290055 |
| [78] |
K.P. Rakesh, S.M. Wang, J. Leng, et al., Anticancer Agents Med. Chem. 18 (2018) 488-505. DOI:10.2174/1871520617666171103140749 |
| [79] |
V.E. Schneeberger, Y. Ren, N. Luetteke, et al., Oncotarget 6 (2015) 6191-6202. DOI:10.18632/oncotarget.3356 |
| [80] |
A. Tomiyama, T. Kobayashi, K. Mori, K. Ichimura, Cancers (Basel) 11 (2019) 241. DOI:10.3390/cancers11020241 |
| [81] |
U.A. Germann, B.F. Furey, W. Markland, et al., Mol. Cancer Ther. 16 (2017) 2351-2363. DOI:10.1158/1535-7163.MCT-17-0456 |
| [82] |
M.J. LaMarche, M. Acker, A. Argintaru, et al., J. Med. Chem. 63 (2020) 13578-13594. DOI:10.1021/acs.jmedchem.0c01170 |
| [83] |
K.P. Papadopoulos, S.H.I. Ou, M.L. Johnson, et al., J. Clin. Oncol. 37 (2019) 3161. DOI:10.1200/JCO.2019.37.15_suppl.TPS3161 |
| [84] |
J. Hallin, L.D. Engstrom, L. Hargis, et al., Cancer Discov. 10 (2020) 54-71. DOI:10.1158/2159-8290.CD-19-1167 |
| [85] |
D. Qian, M. Behera, C.E. Steuer, et al., J. Clin. Oncol. 38 (2020) 21079. DOI:10.1200/JCO.2020.38.15_suppl.e21079 |
| [86] |
H. Lu, C. Liu, H. Huynh, et al., Oncotarget 11 (2020) 265-281. DOI:10.18632/oncotarget.27435 |
| [87] |
C. Bredrup, T. Stokowy, J. McGaughran, et al., Eur. J. Hum. Genet. 27 (2019) 574-581. DOI:10.1038/s41431-018-0323-z |
| [88] |
A.H. Sharpe, K.E. Pauken, Nat. Rev. Immunol. 18 (2018) 153-167. DOI:10.1038/nri.2017.108 |
| [89] |
J. Gong, A. Chehrazi-Raffle, S. Reddi, R. Salgia, J. Immunother. Cancer 6 (2018) 8. DOI:10.1186/s40425-018-0316-z |
| [90] |
M. Li, R. Schulz, M. Chisholm-Burns, J. Wang, Z.K. Lu, J. Manag. Care Spec. Pharm. 26 (2020) 1477-1486. |
| [91] |
C. Kim, B. Kim, Nutrients 10 (2018) 1021. DOI:10.3390/nu10081021 |
| [92] |
J. Du, J. Guo, D. Kang, et al., Chin. Chem. Lett. 31 (2020) 1695-1708. DOI:10.1016/j.cclet.2020.03.028 |