 2021, Vol. 32
2021, Vol. 32 


b Personalized Drug Therapy Key Laboratory of Sichuan Province, Department of Pharmacy, Sichuan Provincial People's Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610072, China
Gambogic acid (GA) is the main active compound of traditional Chinese medicine Gamboge with broad-spectrum anti-inflammatory, antiviral and antitumor activities [1, 2]. It shows strong inhibitory effects on the proliferation of a variety of cancer cells. Moreover, GA can reverse tumor resistance and improve the efficacy of existing chemotherapeutic agents for synergistic cancer treatments [3, 4]. However, the clinical outcome of GA is limited by its poor water solubility and low chemical stability [5]. Currently, a number of formulations have been developed using l-arginine and polyoxylated castor oil as solubilization agents for the intravenous administration of GA, which also suffered from rapid blood clearance and caused some undesirable toxic effects [6, 7]. To address this issue, researchers have incorporated GA into the core of polymeric micelles through hydrophobic interaction [8-10]. Nonetheless, the micellar nanomedicines usually demonstrate unsatisfactory drug loading and nonspecific drug release [11-14]. As an alternative, GA could be covalently conjugated to polymeric structures using its carboxyl group [15-17]. Still, it needs cumbersome chemistry and may compromise the activity of drugs. Therefore, the development of facile and effective GA delivery system is of great significance for effective cancer chemotherapy.
It has been shown that GA can bind to the N-terminal domain of heat shock protein 90 via electrostatic interaction, which causes a conformational change in proteins and plays an important role in exerting its anticancer effects [18, 19]. Inspired by this finding, the complexation of GA and polymers would be a promising strategy for construction of drug delivery systems. As a class of protein-mimicking synthetic polypeptides, poly(amino acid)s (PAA) with excellent biocompatibility and biodegradability can form unique secondary conformations (e.g., α-helix, β-sheet, et al.) as well as hierarchically assembled structures in aqueous solution [20-25]. Moreover, the ionizable side groups of PAA provide abundant active sites for further modification and functionalization, which could also be used to immobilize drugs, proteins and nucleic acids through electrostatic interaction [26-33]. The electrostatic complexation of GA with PAA is advantageous due to its simplicity, specificity, and reversibility, which does not involve toxic agents and complicated reactions. However, the effect of drug complexation on the hierarchical assembly of polymers has not been well understood.
Herein, we report a facile GA-delivery system using a diblock methoxy poly(ethylene glycol)-b-poly(L-lysine) bearing a disulfide linkage (MPEG-SS-PLL) as a drug binding model. GA can be effectively incorporated into the system via electrostatic interaction with high drug loading content. Interestingly, the complexation of drugs had a dramatic impact on the secondary conformations and assembled morphologies (Fig. 1A). The drug-induced self-assembly (DISA) of PAA was investigated in detail, and the multi-stimuli-responsive properties, drug release profiles, cell internalization and synergistic antitumor performance in vitro were evaluated to explore the potential of nanocomplexes in drug delivery applications.

|
Download:
|
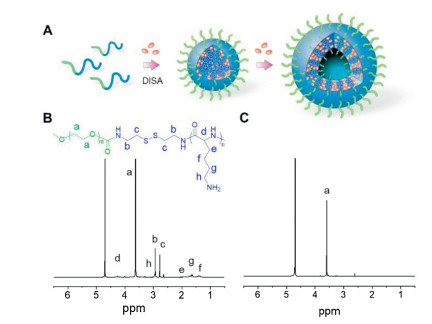
| Fig. 1. (A) Schematic illustration of drug-induced hierarchical self-assembly of poly(amino acid). 400 MHz1H NMR spectra of MPEG-SS-PLL (B) and MPEG-SS-PLL-GA complex (C) in D2O. | |
{kind=link}
To construct the model polymer, a methoxypolyethylene glycol amine bearing a disulfide linkage (MPEG-SS-NH2) and N(ε)-benzyloxycarbonyl-L-lysine N-carboxyanhydride (ZLL-NCA) were firstly synthesized (Schemes S1 and S2 in Supporting information). Then a MPEG-SS-PZLL diblock copolymer was prepared by the ring-opening polymerization (ROP) of ZLL-NCA initiated by MPEG-SS-NH2, followed by deprotection of Z groups in the presence of trifluoroacetic acid (TFA) and HBr/acetic acid to give MPEG-SS-PLL (Supporting information). The resultant copolymer showed a narrow molecular distribution as determined by gel permeation chromatography (GPC) (Fig. S1 in Supporting information). 1H NMR spectra show the characteristic peaks of MPEG, PLL segment, Z groups and cystamine residues (Fig. 1B and Fig. S2 in Supporting information), confirming the successful synthesis of MPEG-SS-PZLL and MPEG-SS-PLL. The double hydrophilic character enables good water solubility of MPEG-SS-PLL. However, the resonance peaks attributed to PLL segment appeared relatively weaker than those of disulfide linkages in D2O (Fig. 1B), suggesting slight association of PLL block in an aqueous solution. Further, the proton signals of PLL segment completely disappeared in the electrostatic complex of MPEG-SS-PLL and GA (MPEG-SS-PLL-GA) (Fig. 1C). The result reveals a restricted motion of protons due to the drug-induced hydrophobic aggregation of PLL block [34]. The self-assembly of the complexed systems was also confirmed by low critical aggregation concentrations (CAC) (10.5–13.4 μg/mL, Fig. S3 in Supporting information), suggesting a high thermodynamic stability of the assemblies.
To verify whether electrostatic complexation imparted a high drug loading capacity, the content of encapsulated GA was determined using UV–vis spectroscopy. Encouragingly, the drug loading content (DLC) increased with increasing of drug feeding ratios and reached up to 53.6 % (Fig. S4 in Supporting information), which was much higher than those for traditional nanoformulations encapsulating GA via hydrophobic interaction (1 %–26 %) [8-10]. Moreover, all the drug-complexed MPEG-SS-PLL assemblies present a GA absorption at 272 nm, much lower than that of free GA (290 nm). The hypsochromic shift verifies effective binding of drugs with polymers and rules out the existence of free GA that led to a redshift of UV absorption (Fig. 2A) [35]. Further high-performance liquid chromatography (HPLC) confirmed the purity of complexes, where only one peak of polymeric assemblies was observed and no signal of free drugs was found (Fig. S5 in Supporting information) [36]. In addition, it is interesting to notice that the turbidity of the mixture increased suddenly as drug/polymer feeding ratio was higher than 0.8 (Fig. 2B), implying a possible drug-induced morphological transition. To understand this phenomenon, the complexes were determined with dynamic light-scattering (DLS) and transmission electron microscopy (TEM). It was found that the particle sizes increased from 33 nm to 187 nm and the morphologies of assemblies changed from micelles to vesicles with an increase in DLC (Fig. 2C, E and F). To further justify the morphological transition, taking assemblies with the lowest and highest DLC as examples, a rhodamine 6G (R6G) encapsulation test was carried out. We found that the nanocomplexes with lower DLC cannot accommodate hydrophilic guest, while those containing more drugs can encapsulate the dyes favorably with a self-quenching of R6G fluorescence (Fig. 2D). The results confirm the drug complexation-induced micelle-to-vesicle transition of polymeric assemblies.

|
Download:
|
| Fig. 2. (A) UV–vis spectra of GA, MPEG-SS-PLL and MPEG-SS-PLL-GA complex in methanol. For comparison, polymeric systems mixed with free GA were tested as controls (MPEG-SS-PLL+GA and MPEG-SS-PLL-GA+GA). (B) Turbidity of MPEG-SS-PLL solutions before and after complexation with GA in different drug feeding ratios. The insets show digital pictures of corresponding solution and drug complexes. (C) Size distributions of MPEG-SS-PLL-GA complexes with different drug feeding ratios: (a) 0.5, (b) 0.8, (c) 1.5, (d) 2. (D) UV–vis spectra (a-c, left) and fluorescence emission spectra (d-f, right) of R6G in water (a, d) and MPEG-SS-PLL-GA complexes with drug feeding ratio of 0.5 (MPEG-SS-PLL-GA0.5) (b, e) and 1.5 (MPEG-SS-PLL-GA1.5) (c, f). The insets show emission images of R6G in water (upper), MPEG-SS-PLL-GA0.5 (g) and MPEG-SS-PLL-GA1.5 (h). TEM images of MPEG-SS-PLL-GA0.5 (E) and MPEG-SS-PLL-GA1.5 (F). The scale bars are 100 nm. | |
{kind=link}
The transformation of morphology was largely due to the increase of hydrophobic volume fraction by GA complexation, which decreased the interfacial curvature between MPEG and PLL domains [37]. On the other hand, the secondary conformations of poly(amino acid)s should also be taken into account, as we and others have shown that the formation of α-helical conformation favors vesicular structure [20, 38]. To clarify this point, we performed circular dichroism (CD) measurements on the assemblies. We found that MPEG-SS-PLL exhibited a random coil structure under neutral condition (Fig. S6), because of charge repulsion between the side chains [39]. However, the analysis of CD spectra of drug-complexed polymers was complicated since GA itself generated strong CD signals. Still, it can be judged that MPEG-SS-PLL-GA adopted an ordered conformation based on the positive band around 193 nm in the CD spectrum (Fig. S6 in Supporting information) [40]. Moreover, the Fourier transform infrared (FTIR) spectra present amide Ⅰ and Ⅱ bands at 1650 and 1544 cm−1, respectively, suggesting an α-helical conformation of MPEG-SS-PLL-GA (Fig. S7 in Supporting information) [41].
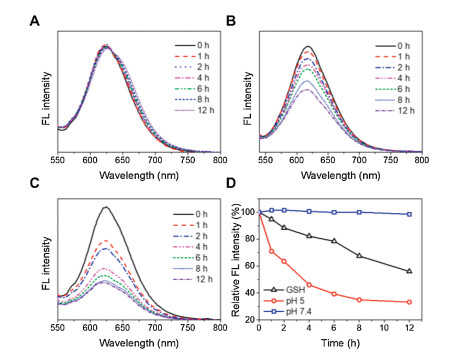
The reversible character of electrostatic complexation allows for the dissociation of drugs and polymers under acidic conditions. As the pH of assemblies decreased to ~5, a lysosome-mimicking environment [42, 43], the UV absorbance of drug complexes declined overtime because GA was protonated and released toward aqueous solution (Fig. S8 in Supporting information). The detachment of GA decreased the hydrophobicity of nanocomplexes and resulted in the rapture of polymer assemblies, which provides an opportunity for controlled delivery of payloads. To test this potential, we incorporated nile red (NR) into the assemblies. The NR-loaded nanocarriers were incubated in different media and monitored with fluorescence spectroscopy. It was found that the NR fluorescence kept unchanged under normal physiological condition (pH 7.4), and decreased significantly in acidic (pH 5) or reductive (10 mmol/L glutathione (GSH)) environments (Fig. 3). Note that the release rate at pH 5 was much higher than that in the presence of GSH. There are probably two reasons to account for this phenomenon. One is that the pH-induced protonation of GA is sensitive with more reactive sites, facilitating a simultaneous release of both drugs. The other reason is that the disulfide bonds linked with PLL segments were distributed near the core during self-assembly, which may render a steric hindrance to the attack of GSH and limit the responsivity of nanosystems [44-46].

|
Download:
|
| Fig. 3. Fluorescence spectra of NR-loaded MPEG-SS-PLL-GA complexes incubated in (A) pH 7.4, (B) 10 mmol/L GSH, and (C) pH 5 for different times. (D) Normalized decrease in fluorescence intensity against time for NR-loaded MPEG-SS-PLL-GA complexes. | |
{kind=link}
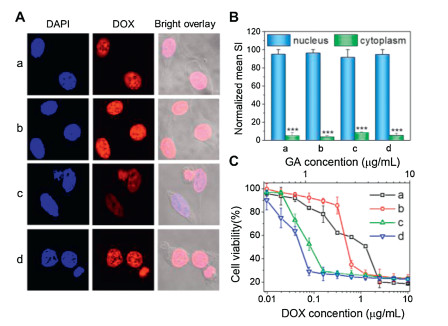
To test the potential of hierarchically assembled drug nanocomplexes in intracellular drug delivery, we incorporated an anticancer drug doxorubicin (DOX) into the assemblies. The encapsulation of DOX did not impact on the particle size, further demonstrating a high stability of complexed systems (Fig. S9 in Supporting information). Interestingly, the nanosystem showed strong DOX fluorescence observed mainly in the cell nuclei, revealing a high cell internalization efficiency comparable to that of free DOX (Fig. 4). This is because their cationic and rigid α-helical structure could interact efficiently with the lipid bilayers for improved cellular entry as well as triggered drug release within tumor cells enabled free DOX to readily infiltrate into the cell nucleus [20, 47-49]. As a result, the nanoformulations exerted potent and synergistic drug efficacy in vitro, with antitumor activity much higher than those of free drugs (Fig. 4C). In addition, the drug-free MPEG-SS-PLL were cytocompatible (Fig. S10 in Supporting information), which could be potentially biodegraded into nontoxic PEG and natural l-amino acids for safe application in vivo.

|
Download:
|
| Fig. 4. (A) CLSM images of MCF-7 tumor cells incubated with DOX (a, b) and DOX-loaded MPEG-SS-PLL-GA complexes (c, d) for 1 h (a, c) and 2 h (b, d). Nuclei of cells were stained with DAPI. (B) Mean SI of DOX fluorescence in the cytoplasm and nucleus calculated and normalized with ImageJ software. (C) Viability of MCF-7 cancer cells incubated with DOX (a), GA (b), MPEG-SS-PLL-GA (c) and DOX-loaded MPEG-SS-PLL-GA complexes (d) at different drug concentrations. | |
{kind=link}
In summary, we have developed a facile GA-complexed polymer system with high drug loading capacity. The GA bound with the side chains of copolymers through electrostatic interaction and induced a conformation transition from random coil to α-helix, combined with a micelle-to-vesicle transformation. The drug-constructed nanocarriers could further accommodate therapeutic agents for efficient cell internalization and specific intracellular drug release, resulting in a synergistic antitumor effect in vitro. The DISA approach is simple and specific, without use of toxic agents and cumbersome chemistry, which can be potentially applied to other polymers and therapeutics for controlled delivery applications.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe research was supported by the National Natural Science Foundation of China (Nos. 51873118, 21474064, 52022062), the Key Research and Development Program of Science and Technology Department of Sichuan Province (No. 2019YFS0514), Science and Technology Project of Chengdu (No. 2019-YF05-00784-SN), the Project of State Key Laboratory of Polymer Materials Engineering (No. sklpme2020-2-03) and the Fundamental Research Funds for Central Universities.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.016.
| [1] |
T. Yi, Z. Yi, S. Cho, et al., Cancer Res. 68 (2008) 1843. DOI:10.1158/0008-5472.CAN-07-5944 |
| [2] |
K. Banik, C. Harsha, D. Bordoloi, et al., Cancer Lett. 416 (2018) 75-86. DOI:10.1016/j.canlet.2017.12.014 |
| [3] |
D. Kashyap, R. Mondal, H.S. Tuli, G. Kumar, A.K. Sharma, Tumor Biol. 37 (2016) 12915-12925. DOI:10.1007/s13277-016-5194-8 |
| [4] |
Z. Chen, G. Hong, Z. Liu, et al., Colloid. Surf. B 196 (2020) 111286. DOI:10.1016/j.colsurfb.2020.111286 |
| [5] |
F. Yu, C. He, A.Y. Waddad, et al., Drug Dev. Ind. Pharm. 40 (2014) 774-782. DOI:10.3109/03639045.2013.786723 |
| [6] |
Y. Zhang, Z. Yang, X. Tan, X. Tang, Z. Yang, AAPS PharmSciTech 18 (2017) 1987-1997. DOI:10.1208/s12249-016-0670-4 |
| [7] |
L. Cai, N. Qiu, M. Xiang, et al., Int. J. Nanomed. 9 (2014) 243-255. |
| [8] |
L. Han, Y. Wang, X. Huang, et al., Biomaterials 257 (2020) 120228. DOI:10.1016/j.biomaterials.2020.120228 |
| [9] |
Y. Wang, X. Wang, J. Zhang, et al., Chin. Chem. Lett. 30 (2019) 885-888. DOI:10.1016/j.cclet.2019.02.018 |
| [10] |
Y. Wang, X. Liang, R. Tong, et al., J. Biomed. Nanotechnol. 14 (2018) 1695-1704. DOI:10.1166/jbn.2018.2626 |
| [11] |
S. Lv, Y. Wu, K. Cai, et al., J. Am. Chem. Soc. 140 (2018) 1235-1238. DOI:10.1021/jacs.7b12776 |
| [12] |
M. He, L. Yu, Y. Yang, et al., Chin. Chem. Lett. 31 (2020) 3178-3182. DOI:10.1016/j.cclet.2020.05.034 |
| [13] |
Y. Liang, Q. Huo, W. Lu, et al., J. Biomed. Nanotechnol. 14 (2018) 1308-1316. DOI:10.1166/jbn.2018.2585 |
| [14] |
C. Hu, F. Fan, Y. Qin, et al., J. Biomed. Nanotechnol. 14 (2018) 2018-2030. DOI:10.1166/jbn.2018.2647 |
| [15] |
R. Ganugula, M. Arora, P. Saini, M. Guada, M.N.V. Ravi Kumar, J. Am. Chem. Soc. 139 (2017) 7203-7216. DOI:10.1021/jacs.6b13231 |
| [16] |
Y. Kang, L. Lu, J. Lan, et al., Acta Biomater. 68 (2018) 137-153. DOI:10.1016/j.actbio.2017.12.028 |
| [17] |
A. Nguyen, H. Ando, R. Böttger, et al., Biomater. Sci. 8 (2020) 4626-4637. DOI:10.1039/D0BM00711K |
| [18] |
L. Zhang, Y. Yi, J. Chen, et al., Biochem. Biophys. Res. Commun. 403 (2010) 282-287. DOI:10.1016/j.bbrc.2010.11.018 |
| [19] |
J. Davenport, J.R. Manjarrez, L. Peterson, et al., J. Nat. Prod. 74 (2011) 1085-1092. DOI:10.1021/np200029q |
| [20] |
H. Liu, R. Wang, J. Wei, et al., J. Am. Chem. Soc. 140 (2018) 6604-6610. DOI:10.1021/jacs.8b01873 |
| [21] |
R.A. Mazo, S. Allison-Logan, F. Karimi, et al., Chem. Soc. Rev. 49 (2020) 4737-4834. DOI:10.1039/C9CS00738E |
| [22] |
Z. Song, Z. Tan, J. Cheng, Macromolecules 52 (2019) 8521-8539. DOI:10.1021/acs.macromol.9b01450 |
| [23] |
X. Zhou, Z. Li, Adv. Healthcare Mater. 7 (2018) 1800020. DOI:10.1002/adhm.201800020 |
| [24] |
C. He, X. Zhuang, Z. Tang, H. Tian, X. Chen, Adv. Healthcare Mater. 1 (2012) 48-78. DOI:10.1002/adhm.201100008 |
| [25] |
W. Hu, M. Ying, S. Zhang, J. Wang, J. Biomed. Nanotechnol. 14 (2018) 1359-1374. DOI:10.1166/jbn.2018.2590 |
| [26] |
J. Beyermann, H. Kukula, Macromolecules 33 (2000) 5906-5911. DOI:10.1021/ma000629m |
| [27] |
A.F. Thünemann, J. Beyermann, C. von Ferber, H. Löwen, Langmuir 16 (2000) 850-857. DOI:10.1021/la9906525 |
| [28] |
A.F. Thünemann, D. Schütt, R. Sachse, H. Schlaad, H. Möhwald, Langmuir 22 (2006) 2323-2328. DOI:10.1021/la0521138 |
| [29] |
T. Govender, S. Stolnik, C. Xiong, et al., J. Control Release 75 (2001) 249-258. DOI:10.1016/S0168-3659(01)00353-4 |
| [30] |
A.F. Thünemann, S. Kubowicz, C. Burger, et al., J. Am. Chem. Soc. 125 (2003) 352-356. DOI:10.1021/ja0277632 |
| [31] |
P. Zheng, Y. Liu, J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1178-1182. DOI:10.1016/j.cclet.2019.12.001 |
| [32] |
S. Lv, Z. Tang, M. Li, et al., Biomaterials 35 (2014) 6118-6129. DOI:10.1016/j.biomaterials.2014.04.034 |
| [33] |
H. Uchida, K. Itaka, T. Nomoto, et al., J. Am. Chem. Soc. 136 (2014) 12396-12405. DOI:10.1021/ja506194z |
| [34] |
M. Ding, N. Song, X. He, et al., ACS Nano 7 (2013) 1918-1928. DOI:10.1021/nn4002769 |
| [35] |
A. Thünemann, Langmuir 13 (1997) 6040-6046. DOI:10.1021/la970756k |
| [36] |
Y. Qu, B. Chu, X. Wei, et al., J. Control Release 296 (2019) 93-106. DOI:10.1016/j.jconrel.2019.01.016 |
| [37] |
J.S. Lee, J. Feijen, J. Control, Release 161 (2012) 473-483. DOI:10.1016/j.jconrel.2011.10.005 |
| [38] |
E.G. Bellomo, M.D. Wyrsta, L. Pakstis, D.J. Pochan, T.J. Deming, Nat. Mater. 3 (2004) 244-248. DOI:10.1038/nmat1093 |
| [39] |
N.P. Gabrielson, H. Lu, L. Yin, et al., Angew. Chem. Int. Ed. 51 (2012) 1143-1147. DOI:10.1002/anie.201104262 |
| [40] |
K. Wang, Y. Liu, C. Li, et al., ACS Macro Lett. 2 (2013) 201-205. DOI:10.1021/mz300568b |
| [41] |
J. Huang, C. Bonduelle, J. Thévenot, S. Lecommandoux, A. Heise, J. Am. Chem. Soc. 134 (2012) 119-122. DOI:10.1021/ja209676p |
| [42] |
C. Weng, N. Fan, T. Xu, et al., Chin. Chem. Lett. 31 (2020) 1490-1498. DOI:10.1016/j.cclet.2019.11.009 |
| [43] |
Z. Shi, Q. Li, L. Mei, Chin. Chem. Lett. 31 (2020) 1345-1356. DOI:10.1016/j.cclet.2020.03.001 |
| [44] |
J. Wei, X. Shuai, R. Wang, et al., Biomaterials 145 (2017) 138-153. DOI:10.1016/j.biomaterials.2017.08.005 |
| [45] |
C. Weng, H. Chen, T. Xu, et al., ACS Mater. Lett. 2 (2020) 602-609. DOI:10.1021/acsmaterialslett.0c00070 |
| [46] |
R. Yang, Y. Zheng, X. Shuai, et al., Adv. Sci. 7 (2020) 1902701. DOI:10.1002/advs.201902701 |
| [47] |
Y. Zheng, C. Weng, C. Cheng, et al., Macromolecules 53 (2020) 5992-6001. DOI:10.1021/acs.macromol.0c00374 |
| [48] |
M. Shi, K. Ho, A. Keating, M.S. Shoichet, Adv. Funct. Mater. 19 (2009) 1689-1696. DOI:10.1002/adfm.200801271 |
| [49] |
J. Liu, X. Ai, H. Zhang, W. Zhuo, P. Mi, J. Biomed. Nanotechnol. 15 (2019) 373-381. DOI:10.1166/jbn.2019.2693 |