 2021, Vol. 32
2021, Vol. 32 


b Department of Neurosurgery, Qilu Hospital and Institute of Brain and Brain-Inspired Science, Cheeloo College of Medicine, Shandong University, Ji'nan 250012, China;
c State Key Laboratory of Microbial Technology, Shandong University, Qingdao 266237, China
Drug delivery systems (DDS) have shown their potential in delivering drugs to tumor sites, reducing side effects, and improving the therapeutic efficacy [1-5]. Metal-organic frameworks (MOFs) composed of metal ions and organic bridging ligands have attracted great attention in biological applications owing to their controlled structure, tunable porosity and functionalization [6-12]. Among the MOF family, zeolitic imidazole framework-8 (ZIF-8) has been regarded as a good candidate of DDS due to the facile preparation approach, low-cytotoxicity, and pH-responsibility [13-19]. However, some issues such as water dispersibility and stability as well as encapsulation and delivery ability still exist [20-23]. In our previous work, poly(ethylene glycol) (PEG) was used as the mineralizer for the scalable synthesis of ZIF-8 nanoparticles (NPs) with tunable sizes, which improved the colloidal dispersibility and stability in aqueous solution as well as the versatility of cargo encapsulation [24]. These unique advantages enable the PEG-mediated ZIF-8 NPs as a promising nanocarrier for controlled drug delivery.
Most of DDS are internalized into cells through the endocytosis pathway [25, 26]. However, when DDS are delivered into cells, encapsulated cargos are typically trapped in endocytic vesicles and accumulated in lysosomes, resulting in reduced therapeutic effects [27, 28]. Thus, the release of cargos from the endosomes/lysosomes is a vital step in therapeutic transportation and tumor treatment [29-31]. To achieve efficient endosomal/lysosomal escape, DDSs modified with toxins, polycations, and photosensitizers have been investigated. However, surface modification with these components could induce immunogenicity and toxicity. DDSs integrated with non-toxic molecules that promote endosomal/lysosomal escape can improve the drug delivery efficacy. Cell penetrating peptides (CPPs), as a class of biomolecules, typically composed of a maximum 30 amino acid residues, are capable to traverse cell membranes. Introducing CPPs into DDS represents a good strategy for enhanced endosomal/lysosomal escape [32, 33]. For example, CPPs containing histidine and arginine (i.e., H4R4) have shown their benefits for endosomal escape and enhanced drug cytotoxicity [34, 35]. To achieve this, functional NPs with stimuli-responsiveness (e.g., pH, redox potential, enzyme and temperature) have been considered as a promising platform for the triggered release of cargos (e.g., CPPs) at the active site [36-40]. Among them, pH-responsive NPs are most attractive due to the significant pH decrease from extracellular microenvironments to endosomes/lysosomes when the NPs are internalized into cells [41-43].
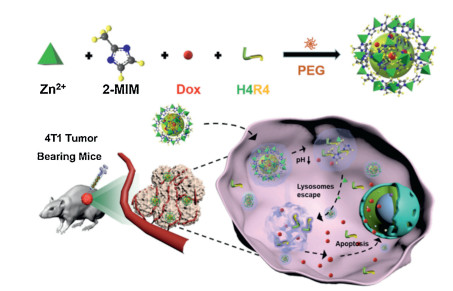
Herein, we reported the preparation of PEG-mediated ZIF-8 NPs for the encapsulation and co-delivery of anticancer drug doxorubicin (Dox) and H4R4 to improve the drug delivery efficacy and inhibit tumor growth (Scheme 1). Cargos were encapsulated in ZIF-8 NPs (Dox & H4R4@ZIF-8 NPs) via a facile one-pot process. The obtained ZIF-8 NPs were stable at physiological condition (e.g., pH 7.4) and could be degradable at acidic microenvironments (e.g., pH 5.5). The reported ZIF-8 NPs offer three advantages for improved drug delivery. First, the DDS was prepared by a facile method without introduction of organic solvents or harsh conditions, which could protect the biological activity of the encapsulated cargos. Second, the obtained NPs were well dispersed in aqueous solution and pH-responsive to control the cargo release. Third, the encapsulated H4R4 without conjugation with Dox could help for endosomal/lysosomal escape to improve the chemotherapeutic efficacy. This reported method could allow different therapeutic molecules to be encapsulated into ZIF-8 NPs and show its great potential in biomedical applications.

|
Download:
|
| Scheme 1. Schematic illustration of preparation of the Dox & H4R4@ZIF-8 NPs to enhance therapeutic delivery. | |
{kind=link}
As a control, Dox and H4R4 were encapsulated separately in ZIF-8 NPs to result in Dox@ZIF-8 and H4R4@ZIF-8 NPs, respectively. The morphology of the obtained NPs was examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). As shown in Fig. 1, ZIF-8, Dox@ZIF-8, H4R4@ZIF-8, Dox & H4R4@ZIF-8 NPs were monodisperse and well-dispersed even at dry state. According to dynamic light scattering (DLS) study, the average hydrodynamic diameter of ZIF-8, Dox@ZIF-8, H4R4@ZIF-8, Dox & H4R4@ZIF-8 NPs in water were 232 nm, 206 nm, 202 nm, 197 nm, respectively (Fig. S1 and Table S1 in Supporting information), consistent with the observation from TEM and SEM measurements.

|
Download:
|
| Fig. 1. (A-D) SEM and (E-H) TEM images of (A, E) ZIF-8, (B, F) Dox@ZIF-8, (C, G) H4R4@ZIF-8 and (D, H) Dox & H4R4@ZIF-8 NPs. Scale bars are 200 nm. | |
{kind=link}
To evaluate the colloidal dispersibility and stability, the NPs were dispersed in water and the hydrodynamic diameter was monitored for eight days using DLS. As a result, the diameter of the NPs did not change significantly, which indicated that the NPs were stable in aqueous solution without further aggregation and demonstrated the excellent colloidal dispersity and stability of our NPs (Fig. 2A). Power X-ray diffraction (PXRD) patterns of the NPs revealed that the crystalline structure of ZIF-8 was well maintained after the encapsulation of Dox and H4R4 peptides (Fig. 2B). Due to the presence of metal ions (i.e., Zn2+), zeta potential of the NPs was positive (~23 mV) as shown in Fig. 2C. However, after incubation with 10% fetal bovine serum solution, zeta potential of the NPs reversed to be negative (−20 mV) due to the adsorption of the negatively charged proteins on the surface of the NPs (Fig. S2 in Supporting information). The loading capacity of Dox and H4R4 in ZIF-8 NPs was 5.2% and 3.7%, respectively, based on calibration curves (Fig. S3 in Supporting information). The loading amount of Dox and H4R4 in ZIF-8 NPs could be tuned by using different concentration of Dox and H4R4. To investigate the pH-responsiveness, Dox@ZIF-8 and Dox & H4R4@ZIF-8 NPs were dispersed in buffers at pH 7.4 and 5.5, and the Dox release was monitored. At pH 7.4, about 20% of Dox was released within 48 h. In contrast, about 90% of the loaded Dox was released from Dox@ZIF-8 NPs at pH 5.5 after 48 h (Fig. 2D). Additionally, Dox & H4R4@ZIF-8 NPs had similar Dox release behavior compared to Dox@ZIF-8 NPs, which indicated that the encapsulation of H4R4 did not influence the drug release. The pH-sensitive disassembly of Dox & H4R4@ZIF-8 NPs was also confirmed by SEM as shown in Fig. S4 (Supporting information), where the NPs were unstable at low pH.

|
Download:
|
| Fig. 2. (A) Hydrodynamic diameters, (B) PXRD patterns, and (C) zeta potential of ZIF-8, Dox@ZIF-8, H4R4@ZIF-8 and Dox & H4R4@ZIF-8 NPs. (D) In vitro release profiles of Dox from Dox@ZIF-8 and Dox & H4R4@ZIF-8 NPs at pH 7.4 and 5.5. | |
{kind=link}
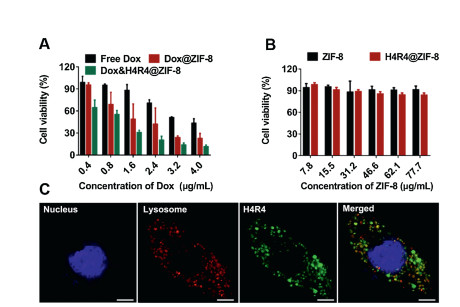
In vitro cell viability of the NPs was evaluated with HeLa and 4T1 cells based on a 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay. As shown in Fig. 3A and Fig. S5 (Supporting information), cell viability of free Dox, Dox@ZIF-8, and Dox & H4R4@ZIF-8 NPs showed dosage-dependent behavior that higher dosage resulted in higher cell cytotoxicity. To demonstrate that the cytotoxicity was due to the encapsulated Dox instead of the NPs, ZIF-8 and H4R4@ZIF-8 NPs with equivalent particle concentration used in Fig. 3A and Fig. S5a were incubated with HeLa and 4T1 cells for 24 h, respectively. As a result, both NPs did not exhibit significant effect on the cell viability, which indicated the biocompatibility of the NPs. Compared with free Dox, Dox@ZIF-8 and Dox & H4R4@ZIF-8 NPs resulted in higher cytotoxicity at an equivalent Dox concentration, which indicated that NPs could improve the drug delivery efficacy. Meanwhile, Dox & H4R4@ZIF-8 NPs induced higher cell apoptosis compared to Dox@ZIF-8 NPs under the same condition, which suggested that the incorporation of H4R4 into NPs led to enhanced cytotoxicity of drug-loaded NPs (Fig. 3B and Fig. S5b). From the cell viability curve, the inhibitory concentration of Dox to produce 50% cell death (IC50) values were 3.22, 1.57, 0.79 μg/mL for free Dox, Dox@ZIF-8 and Dox & H4R4@ZIF-8 NPs, respectively (Table S2 in Supporting information). To investigate the mechanism of the improved drug delivery efficacy, HeLa cells were incubated with H4R4@ZIF-8 NPs and observed by confocal laser scanning microscopy (CLSM). According to the colocalization of lysosomes (red) and fluoresceine isothiocyanate (FITC)-modified H4R4 (green), green signals were observed in cells after 8 h incubation (Fig. 3C and Fig. S6 in Supporting information), which indicated that H4R4 could induce the endosome/lysosome escape and therefore help for the release of Dox from endosomes/lysosomes into cytoplasm to improve the cytotoxicity.

|
Download:
|
| Fig. 3. (A) HeLa cell viability after 24 h incubation with free Dox, Dox@ZIF-8 NPs and Dox & H4R4@ZIF-8 NPs. (B) HeLa cell viability after 24 h incubation with ZIF-8 and H4R4@ZIF-8 NPs (equivalent NP dosages used in A). Data are presented as mean±SD (n=3). (C) CLSM images of HeLa cells after 8 h incubation with H4R4@ZIF-8 NPs. H4R4 was labelled with FITC (green). Nuclei and lysosomes were stained with Hoechst 33258 (blue) and LysoTracker Red (red), respectively. Scale bars are 5 μm. | |
{kind=link}
The acidic microenvironment of lysosomes in HeLa cells could induce the disassembly of ZIF-8 NPs and cargo release. From the CLSM images, in the absence of H4R4, the fluorescence signal of Dox mainly accumulated in the cytoplasm. In contrast, strong red fluorescence signal of Dox was observed in the nucleus of HeLa cells after incubation with Dox & H4R4@ZIF-8 NPs (Fig. S7 in Supporting information). The results further confirmed the flow cytometry data and demonstrated that co-delivery of H4R4 played an important role in facilitating endosomal/lysosomal escape and nuclear translocation of the drug.
To explore the tumor inhibition of Dox & H4R4@ZIF-8 NPs, 4T1 cells were subcutaneously injected into healthy BALB/c female mice to construct 4T1 tumor model. When the tumor size was about 100 mm3, 4T1 tumor-bearing mice were randomly assigned to six groups (n=6) for the injection with PBS, free Dox, ZIF-8, Dox@ZIF-8, H4R4@ZIF-8 and Dox & H4R4@ZIF-8 NPs, respectively. The tumor size and body weight of mice in different groups were recorded every two days. As demonstrated in Fig. 4A, no obvious body weight loss was observed among all groups during the whole treatment. The tumor volumes of mice injected with PBS, free Dox, ZIF-8, Dox@ZIF-8, H4R4@ZIF-8 and Dox & H4R4@ZIF-8 were monitored as well. Compared with PBS group, ZIF-8 and H4R4@ZIF-8 groups did not show significant suppression on tumor growth. Although free Dox and Dox@ZIF-8 NPs exhibited inhibitory effect on tumor growth (free Dox and Dox@ZIF-8 vs. PBS, P < 0.01), enhanced inhibition of tumor size was observed for the group of Dox & H4R4@ZIF-8 (Dox & H4R4@ZIF-8 vs. PBS, P < 0.001) (Fig. 4B). At the end of the treatment, all the mice were sacrificed and the tumors were excised, which further indicated that the mice treated with Dox & H4R4@ZIF-8 NPs resulted in the inhibition of tumor growth (Figs. 4C and D). Compared with the untreated group, hematoxylin-eosin (H & E) staining of major organs demonstrated no obvious damage or inflammatory lesions for the treated groups (Fig. 4E and Fig. S8 in Supporting information), which indicated the low system cytotoxicity of ZIF-8-based delivery systems. Besides, the tumor sections obtained from different groups were stained with Ki-67 and examined using microscope. As shown in Fig. 4E, tumor tissue obtained from the Dox & H4R4@ZIF-8 group showed very few yellow spots, indicating the weak proliferative ability.

|
Download:
|
| Fig. 4. (A) Changes of mouse body weight upon treatments with different drug formulations. (B) Tumor volume changes after post-treatment in 14 days. (C) Average tumor weights after the post-treatment at day 14. (D) Photographs of tumors collected from different treatment groups at day 14. (E) Representative H & E and Ki-67 staining after treatments with PBS and Dox & H4R4@ZIF-8 NPs. Scale bars are 100 μm. *P < 0.05, **P < 0.01 or ***P < 0.001, compared to the PBS group. ##P < 0.01, compared to the free Dox group. | |
{kind=link}
In summary, we fabricated PEG-mediated ZIF-8 NPs for the co-delivery of Dox and H4R4 peptides to improve intracellular drug delivery and tumor inhibition. Due to the pH-responsiveness, ZIF-8 NPs could be disassembled at low pH to release the encapsulated cargos. In vitro results showed that the Dox & H4R4@NPs could improve the drug delivery efficacy via the enhanced endosomal/lysosomal escape. In vivo results indicated Dox & H4R4@ZIF-8 could significantly inhibit the tumor growth while showing minimal system cytotoxicity. The PEG-mediated ZIF-8 NPs with advantages in colloidal stability, encapsulation and delivery ability are promising for improved delivery of therapeutics in biomedical applications.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was supported by the National Natural Science Foundation of China (Nos. 21872085 and 21902088) and the Project for Scientific Research Innovation Team of Young Scholar in Colleges and Universities of Shandong Province (No. 2020KJC001). This work was performed in part at the Translational Medicine Core Facility of Advanced Medical Research Institute at Shandong University.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.011.
| [1] |
D. Rosenblum, N. Joshi, W. Tao, J.M. Karp, D. Peer, Nat. Commun. 9 (2018) 1410. DOI:10.1038/s41467-018-03705-y |
| [2] |
O.S. Fenton, K.N. Olafson, P.S. Pillai, M.J. Mitchell, R. Langer, Adv. Mater. 30 (2018) 1705328. DOI:10.1002/adma.201705328 |
| [3] |
Y. Gao, L. Jia, Q. Wang, et al., ACS Appl. Mater. Interfaces 11 (2019) 16296-16310. DOI:10.1021/acsami.9b02016 |
| [4] |
X. Fu, L.H. Rigau, R. Chandrawati, J. Cui, Chem 4 (2018) 2084-2107. DOI:10.1016/j.chempr.2018.07.002 |
| [5] |
Y. Chen, B. Li, X. Chen, et al., Chin. Chem. Lett. 31 (2020) 1153-1158. DOI:10.1016/j.cclet.2019.06.022 |
| [6] |
L. Zhang, S.S. Wan, C.X. Li, et al., Nano Lett. 18 (2018) 7609-7618. DOI:10.1021/acs.nanolett.8b03178 |
| [7] |
H. Zhang, Q. Li, R. Liu, et al., Adv. Funct. Mater. 28 (2018) 1802830. DOI:10.1002/adfm.201802830 |
| [8] |
H. Zheng, Y. Zhang, L. Liu, et al., J. Am. Chem. Soc. 138 (2016) 962-968. DOI:10.1021/jacs.5b11720 |
| [9] |
M.X. Wu, Y.W. Yang, Adv. Mater. 29 (2017) 1606134. DOI:10.1002/adma.201606134 |
| [10] |
K. Lu, T. Aung, N. Guo, R. Weichselbaum, W. Lin, Adv. Mater. 30 (2018) 1707634. DOI:10.1002/adma.201707634 |
| [11] |
Q. Zhu, M. Saeed, R. Song, et al., Chin. Chem. Lett. 31 (2020) 1051-1059. DOI:10.1016/j.cclet.2019.12.002 |
| [12] |
X. Lai, H. Liu, Y. Zheng, Z. Wang, Y. Chen, J. Biomed. Nanotechnol. 15 (2019) 1754-1763. DOI:10.1166/jbn.2019.2807 |
| [13] |
Y.T. Qin, H. Peng, X.W. He, W.Y. Li, Y. Zhang, ACS Appl. Mater. Interfaces 11 (2019) 34268-34281. DOI:10.1021/acsami.9b12641 |
| [14] |
H. Wang, Y. Chen, H. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 7380-7384. DOI:10.1002/anie.201902714 |
| [15] |
X. Chen, R. Tong, Z. Shi, et al., ACS Appl. Mater. Interfaces 10 (2018) 2328-2337. DOI:10.1021/acsami.7b16522 |
| [16] |
H. Cheng, X.Y. Jiang, R.R. Zheng, et al., Biomaterials 195 (2019) 75-85. DOI:10.1016/j.biomaterials.2019.01.003 |
| [17] |
L. Gao, Q. Chen, T. Gong, J. Liu, C. Li, Nanoscale 11 (2019) 21030-21045. DOI:10.1039/C9NR06558J |
| [18] |
C. Yang, J. Xu, D. Yang, et al., Chin. Chem. Lett. 29 (2018) 1421-1424. DOI:10.1016/j.cclet.2018.02.014 |
| [19] |
S. Zhang, X. Pei, H. Gao, S. Chen, J. Wang, Chin. Chem. Lett. 31 (2020) 1060-1070. DOI:10.1016/j.cclet.2019.11.036 |
| [20] |
S. Liu, C. Hu, Y. Liu, et al., Chem. Eur. J. 25 (2019) 4315-4319. DOI:10.1002/chem.201806242 |
| [21] |
T. Du, Z. Qin, Y. Zheng, et al., Chem 5 (2019) 2942-2954. DOI:10.1016/j.chempr.2019.08.018 |
| [22] |
J. Zhou, M. Li, Y. Hou, et al., ACS Nano 12 (2018) 2858-2872. DOI:10.1021/acsnano.8b00309 |
| [23] |
S. Li, K. Wang, Y. Shi, et al., Adv. Funct. Mater. 26 (2016) 2715-2727. DOI:10.1002/adfm.201504998 |
| [24] |
Q. Yu, Y. Tian, M. Li, et al., Chem. Commun. 56 (2020) 11078-11081. DOI:10.1039/D0CC03734F |
| [25] |
W. He, X. Xing, X. Wang, et al., Adv. Funct. Mater. 30 (2020) 1910566. DOI:10.1002/adfm.201910566 |
| [26] |
H. Xiong, Z. Wang, C. Wang, J. Yao, Nano Lett. 20 (2020) 1781-1790. DOI:10.1021/acs.nanolett.9b05008 |
| [27] |
S.A. Smith, L.I. Selby, A.P. Johnston, G.K. Such, Bioconjug. Chem. 30 (2018) 263-272. |
| [28] |
K. Dong, Z. Wang, Y. Zhang, J. Ren, X. Qu, ACS Appl. Mater. Interfaces 10 (2018) 31998-32005. DOI:10.1021/acsami.8b11972 |
| [29] |
W.Q. Li, L.P. Sun, Y. Xia, et al., ACS Appl. Mater. Interfaces 10 (2018) 5340-5347. DOI:10.1021/acsami.7b18577 |
| [30] |
Y. Liu, C.S. Gong, Y. Dai, et al., Biomaterials 218 (2019) 119365. DOI:10.1016/j.biomaterials.2019.119365 |
| [31] |
G. Lin, Y. Zhang, L. Zhang, et al., Nano Res. 13 (2020) 238-245. DOI:10.1007/s12274-019-2606-2 |
| [32] |
S. Silva, A.J. Almeida, N. Vale, Biomolecules 9 (2019) 22. DOI:10.3390/biom9010022 |
| [33] |
X. Liu, J. Liu, D. Liu, et al., Biomater. Sci. 7 (2019) 5516-5527. DOI:10.1039/C9BM01183H |
| [34] |
K. Liang, J.J. Richardson, H. Ejima, et al., Adv. Mater. 26 (2014) 2398-2402. DOI:10.1002/adma.201305002 |
| [35] |
T. Jiang, R. Mo, A. Bellotti, J. Zhou, Z. Gu, Adv. Funct. Mater. 24 (2014) 2295-2304. DOI:10.1002/adfm.201303222 |
| [36] |
K. Yang, L. Feng, Z. Liu, Adv. Drug Deliv. Rev. 105 (2016) 228-241. DOI:10.1016/j.addr.2016.05.015 |
| [37] |
J.C. Yang, Y. Shang, Y.H. Li, Y. Cui, X.B. Yin, Chem. Sci. 9 (2018) 7210-7217. DOI:10.1039/C8SC02305K |
| [38] |
L. Wang, M. Huo, Y. Chen, J. Shi, Adv. Healthc. Mater. 7 (2018) 1701156. DOI:10.1002/adhm.201701156 |
| [39] |
Y. Li, J. Jin, D. Wang, et al., Nano Res. 11 (2018) 3294-3305. DOI:10.1007/s12274-017-1874-y |
| [40] |
W. Cai, J. Wang, C. Chu, et al., Adv. Sci. 6 (2019) 1801526. DOI:10.1002/advs.201801526 |
| [41] |
Z. Shi, Q. Li, L. Mei, Chin. Chem. Lett. 31 (2020) 1345-1356. DOI:10.1016/j.cclet.2020.03.001 |
| [42] |
P. Zheng, Y. Liu, J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1178-1182. DOI:10.1016/j.cclet.2019.12.001 |
| [43] |
N. Deirram, C. Zhang, S.S. Kermaniyan, A.P.R. Johnston, G.K. Such, Macromol. Rapid Comm. 40 (2019) 1800917. DOI:10.1002/marc.201800917 |