 2021, Vol. 32
2021, Vol. 32 


b Center for Medical Research and Innovation, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai 201399, China
In the past decades, different kinds of nanoparticles have emerged as novel nanocarriers for drug delivery via different administration routes [1-6]. The oral drug delivery route is simple, convenient, economic, painless, and patient-compliant [7, 8]. But it involves a complex absorption process, particularly for the poorly aqueous soluble drugs [9, 10]. In this regard, micelles have been found to enhance drug solubility, stability in the gastrointestinal tract (GIT), mucus penetration, membrane permeability, transport across the intestinal epithelia, absorption and bioavailability, and inhibition of efflux pumps [11-14]. Therefore, it is pivotal to elucidate the mechanisms by which micelles improve the bioavailability of poorly soluble drugs and studies showed that various mechanisms are involved. Enhanced solubility or dissolution and permeability are generally accepted as the leading factors [15-22]. In addition, inhibition of both the P-glycoprotein efflux system and cytochrome P450 metabolism by constituting materials is believed to be one of the underlying mechanisms too [15, 18, 23-25].
Nevertheless, there is always a misunderstanding with regard to mechanisms of micellar solubilization. The micellar systems are commonly regarded as a "solution" of the solutes, but they are actually not. Micelles are first of all particles [26-28], and the loaded drugs are sure to leak or release during transit through the GIT. It is of high significance to elucidate in which form drugs are absorbed-encapsulated in micelles or as free drug molecules. Concerning the applications of micelles in oral drug delivery, previous studies focused mainly on designing new micellar systems and exploring capacities of solubilization. Their use as oral delivery systems has been broadly investigated but not much attention has been paid to their behaviors in the GIT. Prior studies only offered inconclusive results on thein vivo fate of micelles in the GIT and these findings could hardly be employed to elucidate the in vivo fate of micelles. To date, the discrepancies persist and unremittingly urge to explain the oral fate of micelles with corroborative evidence.
The ambiguous understandings about the mechanism of micelles are to some extent due to limitations in discriminating micelle-associated drugs from free drugs. Even the micellar particles themselves are hard to trace. Conventional labeling strategies with radioactive materials or fluorophores suffer from poor discrimination of vehicle-bound signals from free probe signals due to similarity in signals of intact particles and free probes [29, 30]. To address this problem, we employed environmentally responsive water-quenching fluorescent dyes to label micelles [31, 32]. The fluorophores have an aza-4, 4′-difluoro-4-bora-3a, 4a-diaza-s-indacene (aza-BODIPY) parent structure and exhibit absolute aggregation-caused quenching (ACQ) properties. When the dyes are molecularly dispersed in the matrix of nanoparticles, the particles are illuminated. When the dyes are released from the matrix upon degradation of the particles, they aggregate and quench immediately. Therefore, a positive correlation is able to be established between the fluorescence detected and the amount of particles bothin vitro and in vivo. With the help of live imaging tools, the in vivo fate of a series of ACQ dyes-labeled nanocarriers has been investigated [33-44].
Herein, we aim to investigate the intragastrointestinal fate of drug-loaded micelles by tracking the micelles and the model drug paclitaxel (PTX) simultaneously. Eventually, we would like to achieve an explanation for enhanced oral bioavailability of poorly soluble drugs by micelles. To this end, we investigated the in vitro and ex vivo stability of paclitaxel-loaded micelles and drug release, gastrointestinal retention, intestinal epithelial uptake, the potential of micelles in crossing the enterocytes and microfold cells (M cells). We envision that such an investigation would play a role in bridging the gap of previous studies on the fate of micelles and improved bioavailability of poorly soluble drugs by micelles, which moreover highlights the requisite for designing micelles with improved therapeutic efficacy for the oral delivery of poorly soluble drugs.
To track the intragastrointestinal fate of micelles we successfully developed and characterized various fluorescently labeled PTX micelles including Taxol®, d-α-tocopherol polyethylene glycol 1000 succinate (TPGS) and methoxy polyethylene glycol-poly (d, l-lactide) (PEG-PDLLA) micelles. All micellar dispersions were transparent and clear with narrow size distribution. Table S1 (Supporting information) summarizes all micellar formulations and their important characteristics such as size, polydispersity index (PDI), zeta potential, drug loading (DL%) and entrapment efficiency (EE%). Transmission electron microscopy (TEM) analysis revealed the spherical shape and monodispersed size distribution for all micelles formulations (Fig. S1 in Supporting information). Zeta potential for all micelles was near neutral. The EE% for all batches of micelles was above 91% for PTX, exhibiting improved encapsulation due to the lipophilic nature of PTX.
The stability of dye loaded micelles was investigated under both in vitro and ex vivo conditions. For 12h, little variation in fluorescence intensity was observed in water and some other media and fluids exhibiting integral micelles structure, without any obvious dye leakage/soaking by water. Thus, ensuring that dyes can be firmly embedded in micelles, which confirms the precision of follow-up studies.
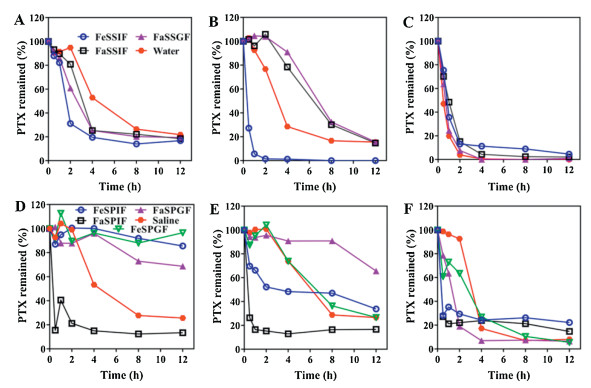
Figs. S2 and S3 (Supporting information) illustrate the stability of micelles in simulated media and pig gastrointestinal fluid, respectively. In vitro stability study showed that all micelles were stable up to 77%–100% in all media except in fed-state simulated intestinal fluid (FeSSIF) for 12h. FeSSIF has high concentrations of surfactants [39] that influenced the micellar stability the most. The effect of material nature is notable in this case. TPGS micelles showed the highest stability in all media except FeSSIF as compared to other groups while PEG-PDDLA micelles stability was a little bit higher in FeSSIF than other groups.
The nature of the material also showed effect on the stability of the micelles inex vivo stability study. In the ex vivo study in 12h, micelles remained integral up to 71%–100% in all fluids except fasted state pig intestinal fluid (FaSPIF) in which 36%–67% micelles disassembled. Quantification of total fluorescence indicated higher stability of TPGS micelles than other groups.
Fig. 1 shows PTX leakage from micelles in simulated and pig gastrointestinal fluid, respectively. Leakage of PTX in 12h for all the formulations in FeSSIF was 83%–100%. The leakage of PTX was intense from all formulations in all media. Apparent PTX precipitation was noted in the drug leakage study. In theex vivo study, in 12h, 83%–87% PTX was released from all formulations in FaSPIF.Ex vivo leakage study also demonstrated fast drug leakage from micelles in a short time in FaSPIF.

|
Download:
|
| Fig. 1. In vitro drug leakage from micelles in simulated media: (A) Taxol®, (B) TPGS and (C) PEG-PDLLA. Ex vivo drug leakage from micelles in pig fluid: (D) Taxol®, (E) TPGS and (F) PEG-PDLLA. The data were presented as percentage of total PTX remained in vehicles by setting the starting concentration to 100%. | |
{kind=link}
Jejunum segment of the intestine was selected for in situ perfusion study to investigate the bioadhesion mechanism of micelles at the gut mucosal surfaces. Comparing different micelles, it was evident that TPGS micelles have higher fluorescence than Taxol® and PEG-PDLLA micelles indicating a good absorption of TPGS in the intestinal segment. Except for TPGS micelles, the difference in absorption of other micelles in the intestinal segment was not very high. This presents the higher association of the TPGS micelles to the epithelial lining as compared to other micelles. The ex vivo images and fluorescence quantification results of rat jejunum segments after first-pass single perfusion of the micelles are shown in Fig. 2.

|
Download:
|
| Fig. 2. Ex vivo imaging of rat jejunum segment after in situ perfusion of micelles (A) and quantification of fluorescence signals of the ex vivo intestinal segment of rat (B). | |
{kind=link}
For the histological investigation of the jejunum segments, confocal laser scanning microscopy (CLSM) was done to detect the P4 signals which were used to indicate the internalization of intact micelles in enterocytes. The results revealed the presence of P4 signals on the outer surfaces of the intestinal microvilli (AP side) with intact micelles permeating to the basolateral (BL) side representing that a fraction of integral micelles were taken up by enterocytes for all formulations (Fig. 3). Strong fluorescence signals of P4/DAPI for TPGS micelles than other groups of micelles confirmed higher uptake of TPGS micelles than Taxol® and PEG-PDLLA micelles being coincident with the results from in situ perfusion. The fluorescence intensity of the PEG-PDLLA micelles was higher than Taxol®.

|
Download:
|
| Fig. 3. CLSM images of rat jejunum frozen section after closed-loop perfusion. DAPI represented in blue, intact micelles represented as red P4 signals and merging of both signals signify the infiltration of the micelles in enterocytes: (A) Taxol®, (B) TPGS and (C) PEG-PDLLA. | |
{kind=link}
Trans-epithelial uptake of micelles in Caco-2 and co-culture models was determined after 4h of incubation at 37 ℃. Figs. S4 and S5 (Supporting information) show the CLSM images of the uptake of intact micelles in Caco-2 as well as co-culture models. CLSM was done either inx-y, x-z or y-z directions to precisely probe the location of intact micelles.
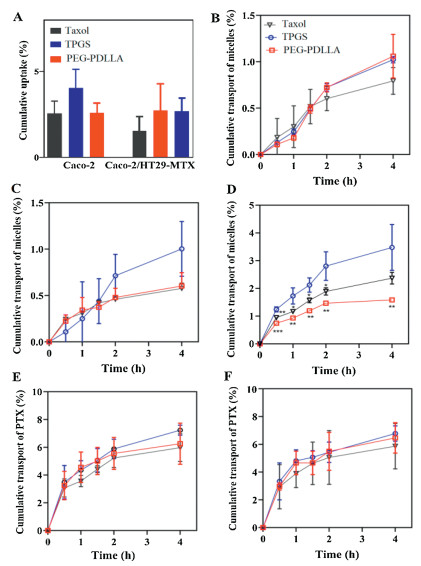
The CLSM images exhibited the uptake of intact micelles in Caco-2 cells, as well as co-culture models, and the fluorescence signals for all micelles irrespective of the nature of materials were scattered throughout the field of view, along with the same horizontal plane with the cell nuclei and in a limited amount at the BL side. A comparison of the uptake of micelles by both cell models showed an insignificant difference. The nature of the polymer showed an impact on the cellular uptake of micelles. Still, the fluorescent signals for TPGS were higher than other groups indicating that TPGS micelles can be internalized in more amount as compared to other micelles groups. The fluorescence intensity was higher for PEG-PDLLA than Taxol®. In a nutshell, as the P4 signals were spotted at both AP and BL side, therefore, it further confirmed the absorption and transport of micelles across the cell monolayers with higher absorption and transport of TPGS micelles.
The uptake of intact micelles was quantitatively measured by fluorescence analysis and it was not apparently different in both cell models but was material dependent (Fig. 4). Uptake of TPGS micelles was higher than other groups in both cell models exhibiting that TPGS micelles were taken up more readily than the other two kinds of micelles. Uptake of PEG-PDLLA was higher than Taxol®.

|
Download:
|
| Fig. 4. Cellular uptake of micelles (A); cumulative transport profiles of micelles vs. time in Caco-2 (B), Caco-2/HT29-MTX (C) and Caco-2/Raji (D) models; cumulative transport profiles of PTX vs. time in Caco-2 (E) and Caco-2/HT29-MTX (F). | |
{kind=link}
TPGS and PEG-PDLLA micelles exhibited about 1% and Taxol® about 0.8% transport of micelles in Caco-2 micelles after 4h. In co-culture cells the transport of TPGS was 1%, 0.6% for PEG-PDLLA and 0.58% for Taxol® after 4h (Fig. 4). Whereas, transport through Caco-/Raji was higher as compared to both cell models and was up to 3.5% for TPGS, 1.6% for PEG-PLLA and 2.4% for Taxol®. The results presented that limited intact micelles were transported across the cell monolayers to the BL side in the Caco-2 and co-culture cell models. These findings exhibited that the trans-monolayer transport of intact micelles by M cells may play a greater part in the oral bioavailability of the drugs than enterocytes but require more corroboration with both imaging as well as quantitative evidence. TPGS micelles transported maximally as intact micelles than other groups. TPGS micelles not only had higher uptake but also remained stable, which was evident by the uptake results. Findings are in coherence with previous results, that is, the material-based absorption of micelles by enterocytes.
The oral drugs require to be transported across the intestinal epithelial barrier, i.e., the first barrier in drug absorption into circulation. So, the transport of PTX was evaluated to investigate the ability of micelles to permeate across the epithelia. The drug encapsulated in micelles was transported across to the BL side. Still, the transport of PTX showed material dependency. The transport of PTX by TPGS was higher than other groups in both cell models. Cumulative transport of PTX by TPGS micelles in Caco-2 cell model was 7.2% while it was 5.9% and 6.2% for Taxol® and PEG-PDLLA, respectively. Cumulative transport of PTX by TPGS micelles in co-culture cell model was 6.8% while it was 5.8% and 6.4% for Taxol® and PEG-PDLLA, respectively. Intact micelles were permeated through the epithelium and PTX was transported. The cumulative transport of PTX was more than the transport of intact micelles through Caco-2 and co-culture cell models which exhibited that most micelles were dissociated to release PTX in cells which was ultimately released in the BL compartment. No difference was measured between the cumulative transport of PTX by Caco-2 and co-culture cell models. Combining the findings of trans-monolayers transport and histological examination of the intestinal segment, it is authenticated that integral micelles penetrated the enteric epithelia and TPGS showed higher penetration.
A concern for oral micellar delivery is that micelles may not be stable under harsh GIT conditions. Although, the micellar system has been designed sophisticatedly; but their ex vivo stability has not been evaluated in the gastrointestinal fluid, consequently, limiting the predictability of the ex vivo investigation to the GIT conditions. Therefore, micelles stability was investigated in both simulated media and ex vivo pig GI fluid. Micelles were treated with pig gastrointestinal fluid to imitate the fate of micelles in the GI harsh conditions during their passage. Stability of micelles may be influenced by fluids with high surfactant and electrolyte contents, e.g. FeSSIF, due to high concentration of phospholipids and bile salts in FeSSIF that can intrude the equilibrium between the unimers and micelles, a similar process that might happen in GIT too. Bile salts and enzymes may notably influence polymeric micelles stability [45], similarly, Taxol® degradation was found to be higher in bile salts than in distilled water [46]. PEG-PDLLA micelles showed excellent stability in the release media without bile salts or enzymes [47]. Micelles remained stable to much extent in all pig GI fluids except FaSPIF. The gastric fluid has less concentration of bile salts and phospholipids than intestinal fluid [48], hence affect the micellar stability less than intestinal fluid. FeSSIF and FaSPIF have affected the stability the most. The discrepancy in the findings ofex vivo and in vitro studies can be ascribed to the reason that simulated media are not true indicators of the GIT conditions. Thus, it can be inferred that micelles stability is materials dependent in gastrointestinal fluid and TPGS micelles exhibited higher stability than Taxol® and PEG-PDLLA micelles despite the generally perceived enhanced stability of polymeric micelles [49-52].
Drug leakage was studied in simulated media and ex vivo pig GI fluid. Fast drug leakage was observed in almost all media particularly FeSSIF and FaSPIF, hence, a rapid drug leakage from micelles would cause the precipitation of the drug in GIT because of the aqueous environment. Taking together, this data also exhibits that micelles will remain assemble but the drug will escape in GIT. Briefly, on oral administration micelles will remain intact but with leakage of the drug prior to absorption.
To investigate the mechanisms at the surface of the intestinal mucosa, retention of micelles in the small intestine was evaluated by in situ perfusion. The perfusion of micelles was studied depending on the fluorescence of the intact micelles and the disappearance of the fluorescence of disassembled micelles in the intestinal lumen. TPGS showed higher bioadhesion than other groups of micelles. Also, from these findings, it can be ascertained that all micelles were internalized in integral form and TPGS showed higher bioadhesion than other groups.
After the nanocarriers are being injected into the intestinal loop carriers may remain in the lumen/trapped by mucus/diffuse through the mucus and enter the epithelial cells. Confocal images of the jejunum segment showed that micelles were found on the villi surface and permeating the BL side, supporting our inference of absorption of intact micelles and reiterating the role of the lymphatic pathway for the intact micelles uptake and transport across the BL surface along with enterocytes. TPGS micelles showed higher uptake than other groups.
The interaction of micelles with epithelium was assessed in Caco-2 and co-culture by confocal visualization to evaluate their ability to permeate the epithelial layer. In both cell models, P4 signals were spotted at both AP and BL side, it further affirmed the transport of intact micelles across the cell monolayers supporting our hypothesis of absorption of intact micelles. All these micelles have also shown uptake previously such as polymeric micelles could cross Caco-2 cells [53] and TPGS can enhance the drug cellular uptake [54]. TPGS micelles exhibited higher internalization than other groups.
The interaction of micelles with gut epithelia was further examined in the Caco-2 and co-culture cell models by fluorescence analysis. The results further proved the uptake of micelles in both cell models. A comparison of uptake of micelles by both cell models showed an insignificant difference and it can be due to the small size of micelles to which the mucus cannot create a barrier to penetration. The commercially available TPGS1000 is so far the most potential efflux pump inhibitor [55] and also in our study TPGS uptake was found higher than other groups in Caco-2 and co-cultures cells. These findings were also evidenced by the results ofin situ intestinal absorption.
Caco-2, co-culture, and Caco-/Raji cell monolayers were utilized to study the transport of PTX and intact micelles. Micelles were permeated and transported across the BL side along with the PTX. Previous studies investigated either the transport of micelles or drug precluding the role of micelles in drug transport. Taxol® was transported across Caco-2 [56], Cremophor EL enhanced the permeability of the P-gp substrate [57]. PEG-PLA has also been reported as a P-gp inhibitor [58]. Studies have shown that TPGS and Cremophor EL have a concentration-dependent inhibition effect [59]. It can be challenging to recognize the most pertinent concentration of preclinical solubilizing agents to imitate conditions employed in oral dosing studies. Nevertheless, a higher amount of micelles was transported across the M cell than Caco-2 and co-culture monolayers. The cumulative transport of PTX was higher than the intact micelles both in Caco-2 and co-culture monolayers which showed the degradation of most micelles and the release of drug in cells and finally released in BL side. The transport of PTX was about same in both cell monolayers. Transport of TPGS and PEG-PDLLA were comparable in Caco-2 cell model and was higher than Taxol®. Transport of TPGS was higher among all formulations in co-culture and Caco-2/Raji cell models. The findings showed that the transcellular transport of intact micelles by M cells may have a pronounced role in the oral bioavailability of the drugs than enterocytes. Both the results of transport and histological examination exhibited the permeation and transport of intact micelles through enteric epithelia. Some previous studies have proved the combination of PEG-PLA/TPGS as a better formulation than PEG-PLA alone. Material dependency was obvious with TPGS showing its potential for better oral absorption. The M cells pathway has a greater role in the absorption of micelles than enterocytes.
This study sought to explore the behavior of micelles in the GIT and their mechanism of absorption and transport. By tracking the signals of various fluorescently labeled micelles, the fate of intact micelles was determined in vitro as well as along the oral passage in ex vivo. It was found that micelles can maintain their integrity in GIT but with prior leakage of PTX. Furthermore, micelles can be absorbed intact through enterocytes and can be transported across the BL side. But the role of M cells in the transport of intact micelles is higher than enterocytes. The nature of material dependency was also confirmed with TPGS showing its potential as a better formulation than other groups. Taking all evidence into account, it is proposed that micelles can be absorbed in an assembled state with prior leakage of the drug in GIT and transported across the intestinal epithelium mainly by M cells and TPGS micelles are better than Taxol® and PEG-PDLLA micelles.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 81872815, 81872826, 82030107, 81690263), and Science and Technology Commission of Shanghai Municipality (No. 19XD1400300).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.09.038.
| [1] |
Y. Qu, B. Chu, X. Wei, et al., J. Control. Release 296 (2019) 93-106. DOI:10.1016/j.jconrel.2019.01.016 |
| [2] |
B. Formicola, A. Cox, R. Dal Magro, et al., J. Biomed. Nanotechnol. 15 (2019) 1997-2004. DOI:10.1166/jbn.2019.2837 |
| [3] |
R. Nayak, I. Meerovich, A.K. Dash, AAPS PharmSciTech 20 (2019) 160. DOI:10.1208/s12249-019-1367-2 |
| [4] |
X. Tang, L. Tan, K. Shi, et al., Acta Pharm. Sin. B 8 (2018) 587-601. DOI:10.1016/j.apsb.2018.05.011 |
| [5] |
X. Niu, J. Chen, J. Gao, Asian J. Pharm. Sci. 14 (2019) 480-496. DOI:10.1016/j.ajps.2018.09.005 |
| [6] |
H. He, Y. Lu, J. Qi, et al., Acta Pharm. Sin. B 9 (2019) 36-48. DOI:10.1016/j.apsb.2018.06.005 |
| [7] |
W. Wu, Y. Lu, J. Qi, Acta Pharm. Sin. B 9 (2019) 2-3. DOI:10.1016/j.apsb.2019.01.009 |
| [8] |
B. Nabi, S. Rehman, S. Baboota, et al., AAPS PharmSciTech 20 (2019) 60. DOI:10.1208/s12249-018-1284-9 |
| [9] |
S. Rani, R. Rana, G.K. Saraogi, et al., AAPS PharmSciTech 20 (2019) 129. DOI:10.1208/s12249-019-1335-x |
| [10] |
H.B. Schultz, T.R. Meola, N. Thomas, et al., Int. J. Pharm. 577 (2020) 119069. DOI:10.1016/j.ijpharm.2020.119069 |
| [11] |
C. Zhu, S. Gong, J. Ding, et al., Acta Pharm. Sin. B 9 (2019) 107-117. DOI:10.1016/j.apsb.2018.09.004 |
| [12] |
V. Piazzini, E. Landucci, M. Urru, et al., Int. J. Pharm. 583 (2020) 119361. DOI:10.1016/j.ijpharm.2020.119361 |
| [13] |
L. Tu, G. Wang, N. Qi, et al., Int. J. Pharm. 578 (2020) 119105. DOI:10.1016/j.ijpharm.2020.119105 |
| [14] |
X. Hou, B. Cao, Y. He, et al., Int. J. Pharm. 569 (2019) 118586. DOI:10.1016/j.ijpharm.2019.118586 |
| [15] |
X. Wang, L. Qiu, X. Wang, et al., Int. J. Pharm. 573 (2020) 118840. DOI:10.1016/j.ijpharm.2019.118840 |
| [16] |
M.N. Bahr, D. Modi, S. Patel, et al., J. Pharm. Pharm. Sci. 22 (2019) 221-246. DOI:10.18433/jpps30347 |
| [17] |
D. Pade, M. Jamei, D.B. Turner, et al., Eur. J. Pharm. Biopharm. 141 (2019) 191-209. DOI:10.1016/j.ejpb.2019.05.024 |
| [18] |
V. Piazzini, M. D'Ambrosio, C. Luceri, et al., Molecules 24 (2019) 1688. DOI:10.3390/molecules24091688 |
| [19] |
J. Wang, L. Wang, Y. Li, et al., Int. J. Nanomed. 13 (2018) 7997. DOI:10.2147/ijn.s183796 |
| [20] |
T. Yin, Y. Zhang, Y. Liu, et al., Int. J. Pharm. 536 (2018) 231-240. DOI:10.1016/j.ijpharm.2017.11.034 |
| [21] |
J. Priotti, D. Leonardi, G. Pico, et al., AAPS PharmSciTech 19 (2018) 1152-1159. DOI:10.1208/s12249-017-0927-6 |
| [22] |
L.L. Wang, D.D. He, S.X. Wang, et al., Drug Dev. Ind. Pharm. 44 (2018) 563-569. DOI:10.1080/03639045.2017.1405431 |
| [23] |
F.Z. Dahmani, H. Yang, J. Zhou, et al., Eur. J. Pharm. Sci. 47 (2012) 179-189. DOI:10.1016/j.ejps.2012.05.015 |
| [24] |
Y. Mu, Y. Fu, J. Li, et al., Carbohydr. Polym. 203 (2019) 10-18. DOI:10.1016/j.carbpol.2018.09.020 |
| [25] |
B. Sharif Makhmal Zadeh, G. Esfahani, A. Salimi, Molecules 23 (2018) 1904. DOI:10.3390/molecules23081904 |
| [26] |
H. He, J. Zhang, Y. Xie, et al., Mol. Pharm. 13 (2016) 4013-4019. DOI:10.1021/acs.molpharmaceut.6b00705 |
| [27] |
Y. Ma, H. He, W. Fan, et al., ACS Biomater. Sci. Eng. 3 (2017) 2399-2409. DOI:10.1021/acsbiomaterials.7b00380 |
| [28] |
L. van Zuylen, J. Verweij, A. Sparreboom, Invest. New Drugs 19 (2001) 125-141. DOI:10.1023/A:1010618632738 |
| [29] |
X. Hu, X. Dong, Y. Lu, et al., Drug Discov. Today 22 (2017) 382-387. DOI:10.1016/j.drudis.2016.10.002 |
| [30] |
W. Wu, T. Li, Adv. Drug Deliv. Rev. 143 (2019) 1-2. DOI:10.23919/aces48530.2019.9060453 |
| [31] |
J. Qi, X. Hu, X. Dong, et al., Adv. Drug Del. Rev. 143 (2019) 206-225. DOI:10.1016/j.addr.2019.05.009 |
| [32] |
X. Hu, J. Zhang, Z. Yu, et al., Nanomedicine 11 (2015) 1939-1948. DOI:10.1016/j.nano.2015.06.013 |
| [33] |
H. He, Y. Xie, Y. Lv, et al., Adv. Healthc. Mater. 7 (2018) 1800711. DOI:10.1002/adhm.201800711 |
| [34] |
H. He, S. Jiang, Y. Xie, et al., Nanoscale Horiz. 3 (2018) 397-407. DOI:10.1039/C8NH00010G |
| [35] |
Y. Xie, B. Shi, F. Xia, et al., J. Control. Release 270 (2018) 65-75. DOI:10.1016/j.jconrel.2017.11.046 |
| [36] |
Y. Ma, H. He, F. Xia, et al., Nanomedicine 13 (2017) 2643-2654. DOI:10.1016/j.nano.2017.07.014 |
| [37] |
F. Xia, W. Fan, S. Jiang, et al., ACS Appl. Mater. Interfaces 9 (2017) 21660-21672. DOI:10.1021/acsami.7b04916 |
| [38] |
E. Ahmad, Y. Feng, J. Qi, et al., Nanoscale 9 (2017) 1174-1183. DOI:10.1039/C6NR07581A |
| [39] |
X. Hu, W. Fan, Z. Yu, et al., Nanoscale 8 (2016) 7024-7035. DOI:10.1039/C5NR07474F |
| [40] |
D. Liu, B. Wan, J. Qi, et al., Chin. Chem. Lett. 29 (2018) 1834-1838. DOI:10.1016/j.cclet.2018.11.015 |
| [41] |
Y. Li, C. Wang, S. Zong, et al., J. Biomed. Nanotechnol. 15 (2019) 686-702. DOI:10.1166/jbn.2019.2724 |
| [42] |
E. Ahmad, Y. Lv, Q. Zhu, et al., Appl. Mater. Today 19 (2020) 100556. DOI:10.1016/j.apmt.2020.100556 |
| [43] |
J. Yang, Z. Dong, W. Liu, et al., Chin. Chem. Lett. 31 (2020) 875-879. DOI:10.1109/lsp.2020.2991357 |
| [44] |
C. Shen, Y. Yang, B. Shen, et al., Nanoscale 10 (2018) 436-450. DOI:10.1039/C7NR06052A |
| [45] |
S.M. Simões, A.R. Figueiras, F. Veiga, et al., Expert Opin. Drug Deliv. 12 (2015) 297-318. DOI:10.1517/17425247.2015.960841 |
| [46] |
H. Montaseri, K.S.F. Jamali, J.A. Rogers, et al., Iran. J. Pharm. Sci. 1 (2005) 43-51. |
| [47] |
E. Pierri, K. Avgoustakis, J. Biomed. Mater. Res. 75 (2005) 639-647. DOI:10.1002/jbm.a.30490 |
| [48] |
P.N. Wiecinski, K.M. Metz, A.N. Mangham, et al., Nanotoxicology 3 (2009) 202-214. DOI:10.1080/17435390902859556 |
| [49] |
S. Shan, Y. Liang, J. Yao, et al., J. Biomed. Nanotechnol. 15 (2019) 674-685. DOI:10.1166/jbn.2019.2721 |
| [50] |
J. Liu, X. Ai, H. Zhang, et al., J. Biomed. Nanotechnol. 15 (2019) 373-381. DOI:10.1166/jbn.2019.2693 |
| [51] |
J. Xie, Y. Lu, B. Yu, et al., Chin. Chem. Lett. 31 (2020) 1173-1177. DOI:10.1016/j.cclet.2019.10.030 |
| [52] |
P. Zheng, Y. Liu, J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1178-1182. DOI:10.1016/j.cclet.2019.12.001 |
| [53] |
F. Mathot, L. van Beijsterveldt, V. Préat, et al., J. Control. Release 111 (2006) 47-55. DOI:10.1016/j.jconrel.2005.11.012 |
| [54] |
Z. Zhang, S. Tan, S.S. Feng, Biomaterials 33 (2012) 4889-4906. DOI:10.1016/j.biomaterials.2012.03.046 |
| [55] |
E.M. Collnot, C. Baldes, M.F. Wempe, et al., J. Control. Release 111 (2006) 35-40. DOI:10.1016/j.jconrel.2005.11.005 |
| [56] |
U.K. Walle, T. Walle, Drug Metab. Dispos. 26 (1998) 343-346. |
| [57] |
B.D. Rege, J.P. Kao, J.E. Polli, Eur. J. Pharm. Sci. 16 (2002) 237-246. DOI:10.1016/S0928-0987(02)00055-6 |
| [58] |
L. Xiao, X. Xiong, X. Sun, Biomaterials 32 (2011) 5148-5157. DOI:10.1016/j.biomaterials.2011.03.071 |
| [59] |
Z. Fan, J. Wu, X. Fang, et al., Int. J. Pharm. 445 (2013) 141-147. DOI:10.1016/j.ijpharm.2013.01.070 |