 2021, Vol. 32
2021, Vol. 32 


Acyclic diterpene dimers (ADDs), a kind of rare natural products originating from acyclic diterpenes with multiple double bonds, were first discovered from the Meliaceae plant Aphanamixis grandifolia by our research group in 2013 [1]. After that, ADDs have attracted considerable attentions and 25 dimers [1-4] and 26 monomers [5-12] with diverse biological activities were isolated from the Aphanamixis genus, and of these, the biomimetic synthesis of dimers aphadilactones A-D was accomplished [13]. To date, the reported ADDs were proposed to be formed via a [4+2] or [2+2] cycloaddition reaction [1-4]. Moreover, acyclic diterpenes can be assembled with other physiologically active substances, for example ascorbic acid (vitamin C), to provide structurally fantastic and biologically important derivatives [14]. Considering the rich double bonds in monomer precursors [5-12], the diversity of location and pattern in polymerisation might exist. As a new structure family [15], their significant anti-inflammatory activities [1, 2] and structural diversities [1-4] stimulated our further investigation of diterpene dimers from the Aphanamixis plants.
In plant extract, these minor low-polar ADDs coexisted with a large quantity of fatty components [1-4], which increased the difficulty of discovery. Comparing their characteristic methyl and double bond structure fragments distinguished from those of fatty acids, NMR-guided investigation of ADDs in seeds of A. polystachya, a timber tree mainly distributed in the tropical and subtropical of Asia [16], was conducted and led to the isolation of three pairs of dimeric acyclic diterpene enantiomers, aphamines A-C (1-3) (Fig. 1). Different from previously reported dimers [1-4], compounds 1-3 bore an unprecedented carbon skeleton through the linkage of a 4H-furan moiety fused via Claisen rearrangement, cyclization and reduction reactions. Herein, the details of NMR-guided isolation, structural elucidation, plausible biosynthetic pathway, and biological activities of 1-3 are described.

|
Download:
|
| Fig. 1. Structures of compounds 1-3. | |
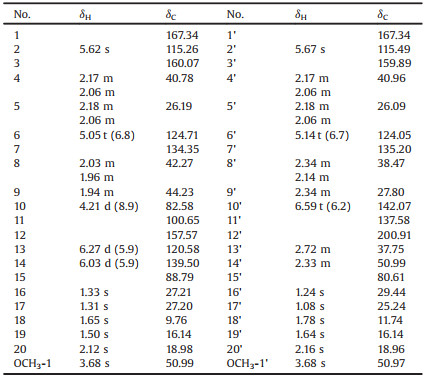
Aphamine A (1), obtained as a colourless gum, had the molecular formula of C42H62O7 determined by its HRESIMS ion at m/z 701.4384 [M+Na]+ (calcd. for 701.4388). The 1H and 13C NMR data (Table 1), interpreted with the help of DEPT and HSQC experiments, revealed resonances for 42 carbons including 12 singlet methyl carbons (two methoxy), eight methylenes, 10 methines (seven olefinic and one oxygenated), nine tertiary carbons (seven olefinic and two oxygenated), and one ketone and two ester carbonyls, of which three resonances appearing highly overlapped (δC 167.34×2, 50.99×2, 16.14×2) and seven ones appearing in pairs (δC 160.07/159.89, 115.49/115.26, 50.99/50.97, 40.96/40.78, 27.21/27.20, 26.19/26.09, 18.98/18.96). Considering the number of methyl singlets and carbon atoms and 12 indices of hydrogen deficiency, compound 1 might be a diterpene dimer similar to the previously described aphanamene A [1].
|
|
Table 1 1H (600MHz) and13C NMR (150MHz) spectroscopic data for compound 1 in CDCl3 (δ in ppm, J in Hz). |
{kind=link}
A 2, 2-dimethyl-2, 5-2 H-furan moiety and one double bond between C-11 and C-12 were established by the HMBC cross-peaks (Fig. 2) of H3-18 (δH 1.65) to C-11 (δC 100.65) and C-12 (δC 157.57); of H-13 (δH 6.27) to C-12, C-14 (δC 139.50), and C-15 (δC 88.79); of H-14 (δH 6.03) to C-12, C-13 (δC 120.58), and C-15; and of H3-16 (δH 1.33) and H3-17 (δH 1.31) to C-14 and C-15. The presence of an α, β-unsaturated-δ-methyl ester moiety was confirmed by the HMBC correlations of OCH3 (δH 3.68) to C-1 (δC 167.34); of H-2 (δH 5.62) to C-1 and C-3 (δC 160.07); and of H3-20 (δH 2.12) to C-1, C-2 (δC 115.26), and C-3. The connection of these above fragments constructed the monomeric diterpene unit A of 1 by the HMBC correlations shown in Fig. 2. In the same way, the monomer unit B was readily established, with the major difference being the absence of the 2, 5-2 H-furan moiety in unit A and the presence of an α, β-unsaturated carbonyl fragment in unit B. This assignment was supported by the HMBC cross-peaks from H-10' (δH 6.59) to C-9' and C-12' (δC 200.91); from H3-18' (δH 1.78) to C-10' (δC 142.07), C-11' (δC 137.58), and C-12'; and from H2-13' to C-12'. The above analysis revealed that both monomer units A and B were acyclic diterpene derivatives derived from melidianolic acid A [5].

|
Download:
|
| Fig. 2. Key HMBC and ROESY correlations of compound 1. | |
{kind=link}
The key HMBC correlations of H2-8 to C-14' (δC 50.99); of H-9 to C-13' (δC 37.75) and C-14'; of H-10 to C-14'; of H-14' to C-8 (δC 42.27) and C-9 (δC 44.23); and of H2-13' to C-9 revealed that C-9 was attached to C-14' through a carbon-carbon single bond. The HMBC cross-peaks of H-10 (δH 4.21) to C-9 and C-11; of H-9 (δH 1.94) and H3-18 to C-10 (δC 82.58); and of H-14' (δH 2.33), H3-16', and H3-17' to C-15' (δC 80.61), suggested that both C-10 and C-15' were oxygenated. The molecular formula of 1 and the remaining one degree of unsaturation required the linkage of C-10 and C-15' via an oxygen atom. Thus, the 2D structure of 1 was determined to be a diterpene dimer skeleton through the linkage of a 4H-furan moiety.
The ROESY cross-peaks from H-10 (δH 4.21) to H-8 (δH 1.96), H-14' (δH 2.33), and H3-16' (δH 1.24) and from H-14' to H3-16' (Fig. 2) indicated that these protons and Me-16' were cofacial and randomly assigned as α-oriented. The coupling constant (J=8.9Hz) between H-9 and H-10 and ROESY correlations of H-9 (δH 1.94)/H2-13' (δH 2.72) and H2-13'/H3-17' (δH 1.08) suggested that H-9, H2-13', and Me-17' were cofacial and β-oriented. Consequently, the α-oriented long wing at C-9 and β-oriented head incorporating a 2, 5-2 H-furan moiety at C-10 were assigned as trans-oriented, and another long wing at C-14' was fixed as β-oriented. The E-geometry of Δ6, 7, Δ11, 12, Δ6', 7', and Δ10', 11' double bonds were deduced from the ROESY correlations of H-6/H-8, H-10/H-13, H-6'/H-8', H-9'/H3-18', and H-10'/H2-13', while the ROESY cross-peaks of H-2/H3-20 and H-2'/H3-20' suggested the Z-geometry of Δ2, 3 and Δ2', 3' double bonds.
The lack of optical activity and Cotton effect suggested that 1 might be a racemic mixture. Chiral HPLC separation of 1 yielded two enantiomers, (+)-1 and (-)-1, in a rate of 1:1 with mirror-image ECD spectra (Figs. 3a and Fig. 3b) and opposite optical rotations. Due to the oil property and flexible structure of acyclic diterpene dimers [1-4], the recrystallization of (+)-1 and (-)-1 in many solvent systems and temperature conditions was unsuccessful. Thus, the absolute configurations of these enantiomers were established by exciton chirality and ECD calculation. The ECD spectrum of (+)-1 (Fig. 3b) showed a positive Cotton effect at 274 nm (Δε +3.94) attributable to n-π* band of the conjugated dienol moiety and a negative Cotton effect at 218 nm (Δε -1.83) assignable to π-π* band of the α, β-unsaturated carbonyl group, indicating a clockwise spatial relationship of these two chromophores [17]. Therefore, the absolute configuration of (+)-1 was defined as 9S, 10S and 14'S. The high similarity of the experimental and calculated ECD data (Fig. 3b) further confirmed the above assignment. Consequently, the opposite CD curve of (-)-1 versus that of (+)-1 revealed the reversed configuration (9R, 10R and 14'R) of (-)-1.

|
Download:
|
| Fig. 3. The chiral HPLC separation of compounds 1 and 3, the experimental and calculated ECD spectra of 1 and 3 in MeOH, and the exciton chirality of the core part of (+)-1. | |
{kind=link}
Comparison of the 1H and 13C NMR data of aphamine B (2) with those of 1 revealed the absence of a methoxy group. The minor variations of the chemical shifts of C-1'/2'/3' (Table S1 in Supporting information) and HMBC cross-peaks of H-2' (δH 5.69) to C-1' (δC 169.53) and C-3' (δC 162.46) and of H3-20' (δH 2.17) to C-1', C-2' (δC 114.89), and C-3' (Fig. S2 in Supporting information) verified the presence of an α, β-unsaturated carboxylic acid group in the unit B of 2 rather than the α, β-unsaturated-δ-methyl ester moiety in 1. The relative configuration of 2 was determined to be the same as that of 1 by a ROESY experiment and the identical chemical shifts of protons and carbons around the 4H-furan ring.
Detailed analysis of the HRESIMS, 1D NMR, HSQC, and HMBC data indicated that aphamine C (3) possessed the same 2D structure as that of 1. The chemical shifts (δH 2.61 (H-9), 4.31 (H-10), 2.60 (H-14'); δC 40.27 (C-9), 82.38 (C-10), 46.76 (C-14'), 81.70 (C-15')) of the protons and carbons around the 4 H-furan ring (Table S1 in Supporting information) were notably different from those (δH 1.94 (H-9), 4.21 (H-10), 2.33 (H-14'); δC 44.23 (C-9), 82.58 (C-10), 50.99 (C-14'), 80.61 (C-15')) in 1, implying that they might be stereoisomers with different configuration at C-9, 10, and 14'. The ROESY cross-peaks of H-8 (δH 1.90)/H-10 (δH 4.31), H-8/H2-13' (δH 2.63, 2.67), H2-13'/H3-16' (δH 1.10), and H3-16'/H-10 (Fig. S3 in Supporting information) revealed that these protons and Me-16 were cofical and assigned as α-oriented. The β-orientations of H-9, H-14', and Me-17' were inferred from the ROESY correlations of H-9 (δH 2.61)/H3-17' (δH 1.26) and H-14' (δH 2.60)/H3-17' and the coupling constant (J9, 10=6.6Hz). Consequently, the long wing at C-9 and the head at C-10 were also trans-oriented, identical to that in 1, while the long wing at C-14' in 3 was determined as α-oriented. Therefore, 3 was a stereoisomer of 1 with different relative configuration at C-14'.
Aphamines B and C (2 and 3) were also resolved by chiral HPLC to obtain (+)-2 and (-)-2 and (+)-3 and (-)-3 (Fig. 3c and Fig. S5 in Supporting information). Similarly, the absolute configurations of (+)-2, (-)-2, (+)-3 and (-)-3 were assigned as 9S, 10S and 14'S; 9R, 10R and 14'R; 9S, 10S and 14'R; and 9R, 10R and 14'S, respectively, based on their experimental and calculated ECD data (Fig. 3d and Fig. S5).
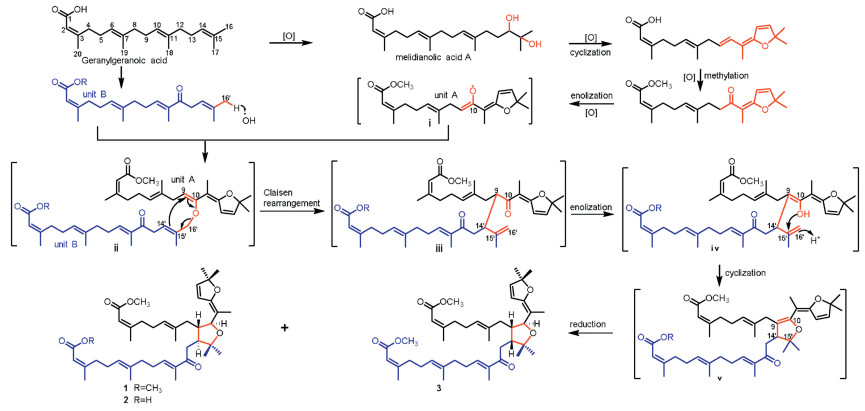
The biogenetic precursor of 1-3 was traced to geranylgeranoic acid, which produced the monomer units A and B through oxidation and cyclization (Scheme 1). Different from previously reported dimers formed via a [4+2] or [2+2] cycloaddition [1-4], oxidation of the olefinic alcohol moiety in unit A would generate a free radical ⅰ, which would be captured by unit B to give intermediate ⅱ [14]. Subsequent Claisen rearrangement of ⅱ provided the intermediate ⅲ [14, 18-20], which then underwent an intramolecular 5-exo-trig cyclization to produce the intermediate ⅴ [14, 21-23]. The asymmetric reduction of Δ9, 10 double bond in ⅴ would afford 1-3 with the wing at C-9 and the head at C-10 in a trans-orientation [24-26]. Based on the aforementioned speculation, the dimeric carbon frameworks of 1-3 represent the third polymerisation mode of acyclic diterpenes.

|
Download:
|
| Scheme 1. Plausible biogenetic pathway of compounds 1-3. | |
{kind=link}
Inflammation in humans is associated with the pathogenesis of various diseases such as cancer, myocardial infraction, rheumatoid arthritis, stroke, and diabetes [27, 28]. Our previous research indicated that 16 diterpene dimers showed significant anti-inflammatory activities [1, 2]. Therefore, compounds 1 and 3 were evaluated for inhibitory activities on NO production in LPS-induced RAW264.7 macrophages [29]. At non-cytotoxic concentrations, compounds (+)-1, (-)-1, (+)-3 and (-)-3 showed inhibitory effects with IC50 values of 11.96±1.08, 9.01±0.53, 15.36±1.11 and 6.71±0.12 μmol/L, respectively (L-NMMA, IC50=43.19±1.99 μmol/L), and reduced the protein level of inducible nitric oxide synthase (iNOS) but had no impact on the expression of cyclooxygenase-2 (COX-2) (Fig. 4 and Fig. S6 in Supporting information).

|
Download:
|
| Fig. 4. The effects of compounds (-)-1 and (-)-3 on NO production and the expression of iNOS and COX-2 in protein levels in LPS-stimulated RAW 264.7 cells. | |
{kind=link}
In conclusion, aphamines A-C (1-3), three pairs of enantiomers, represent a new class of acyclic diterpene dimers bearing a unique carbon skeleton. This finding revealed a new dimerization pattern of acyclic diterpenes involving Claisen rearrangement, 5-exo-trig cyclization, and asymmetric reduction reactions. Meanwhile, their significant inhibitory activities on NO production and reduction of the expression of iNOS provided more evidence for the acyclic diterpene dimers as the candidates for anti-inflammatory agents.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsFinancial support for this study is from the National Natural Science Foundation of China (No. 31470416), the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT_15R63), the "Double First-Class" University Project China (No. CPU2018GY08), the 111 Project from Ministry of Education of China and the State Administration of Foreign Export Affairs of China (No. B18056), and the National New Drug Innovation Major Project (No. 2018ZX09711-001-007).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.028.
| [1] |
H.J. Zhang, J. Luo, S.M. Shan, et al., Org. Lett. 15 (2013) 5512-5515. DOI:10.1021/ol402516p |
| [2] |
H.J. Zhang, Y.M. Zhang, J.G. Luo, J. Luo, L.Y. Kong, Org. Biomol. Chem. 13 (2015) 7452-7458. DOI:10.1039/C5OB00674K |
| [3] |
J. Liu, X.F. He, G.H. Wang, et al., J. Org. Chem. 79 (2014) 599-607. DOI:10.1021/jo402340h |
| [4] |
H. Zhang, J. Liu, L.S. Gan, et al., Org. Biomol. Chem. 14 (2016) 957-962. DOI:10.1039/C5OB02296G |
| [5] |
A. Astulla, Y. Hirasawa, A. Rahman, et al., Nat. Prod. Commun. 6 (2011) 323-326. |
| [6] |
Y. Zhang, J.S. Wang, D.D. Wei, et al., J. Nat. Prod. 76 (2013) 1191-1195. DOI:10.1021/np400126q |
| [7] |
H.F. Wu, X.P. Zhang, Y. Wang, et al., Fitoterapia 90 (2013) 126-131. DOI:10.1016/j.fitote.2013.07.008 |
| [8] |
R. Zhang, H.P. He, Y.T. Di, et al., Fitoterapia 92 (2014) 100-104. DOI:10.1016/j.fitote.2013.10.014 |
| [9] |
X.P. Zhang, Y.F. Tan, Y.B. Li, et al., Chem. Pharm. Bull. 62 (2014) 494-498. DOI:10.1248/cpb.c14-00056 |
| [10] |
H.Y. Zhang, C.M. Yuan, M.M. Cao, et al., Phytochem. Lett. 8 (2014) 81-85. DOI:10.1016/j.phytol.2014.02.005 |
| [11] |
F.H. Fang, W.J. Huang, S.Y. Zhou, et al., Eur. J. Org. Chem. 23 (2017) 4429-4433. |
| [12] |
S. Xue, P.P. Zhang, P.F. Tang, et al., Fitoterapia 142 (2020) 104518. DOI:10.1016/j.fitote.2020.104518 |
| [13] |
J.P. Yin, M. Gu, Y. Li, F.J. Nan, J. Org. Chem. 79 (2014) 6294-6301. DOI:10.1021/jo501117k |
| [14] |
J.X. Zhao, Y.Y. Yu, S.S. Wang, et al., J. Am. Chem. Soc. 140 (2018) 2485-2492. DOI:10.1021/jacs.7b10135 |
| [15] |
G.X. Yang, G.L. Ma, H. Li, et al., Chin. J. Nat. Med. 16 (2018) 881-906. |
| [16] |
S.K. Chen, B.Y. Chen, H. Li, Flora Reipublicae Popularis Sinicae (Zhongguo Zhiwu Zhi). Beijing: Science Press, 1997.
|
| [17] |
N. Berova, L.D. Bari, G. Pescitelli, Chem. Soc. Rev. 36 (2007) 914-931. DOI:10.1039/b515476f |
| [18] |
J.H. Reed, P.A. Donets, S. Miaskiewicz, N. Cramer, Angew. Chem. Int. Ed. 58 (2019) 8893-8897. DOI:10.1002/anie.201904411 |
| [19] |
A.O. Olabisi, M.P.D. Mahindaratne, K. Wimalasena, J. Org. Chem. 70 (2005) 6782-6789. DOI:10.1021/jo0508550 |
| [20] |
K. Wimalasena, M.P.D. Mahindaratne, J. Org. Chem. 59 (1994) 3427-3432. DOI:10.1021/jo00091a036 |
| [21] |
Y. Shi, X.L. Jiang, W.S. Tian, J. Org. Chem. 82 (2017) 269-275. DOI:10.1021/acs.joc.6b02391 |
| [22] |
D. François, M.C. Lallemand, M. Selkti, et al., Angew. Chem. Int. Ed. 37 (1998) 104-105. DOI:10.1002/(SICI)1521-3773(19980202)37:1/2<104::AID-ANIE104>3.0.CO;2-K |
| [23] |
T.G. LaCour, C.X. Guo, S. Bhandaru, M.R. Boyd, P.L. Fuchs, J. Am. Chem. Soc. 120 (1998) 692-707. DOI:10.1021/ja972160p |
| [24] |
R. Bigler, K.A. Mack, J. Shen, et al., Angew. Chem. Int. Ed. 132 (2020) 2866-2871. DOI:10.1002/ange.201912640 |
| [25] |
S. Kraft, K. Ryan, R.B. Kargbo, J. Am. Chem. Soc. 139 (2017) 11630-11641. DOI:10.1021/jacs.7b07188 |
| [26] |
M. Biosca, E. Salomo', P. de la Cruz-Sa'nchez, et al., Org. Lett. 21 (2019) 807-811. DOI:10.1021/acs.orglett.8b04084 |
| [27] |
C.N. Serhan, N.A. Petasis, Chem. Rev. 111 (2011) 5922-5943. DOI:10.1021/cr100396c |
| [28] |
F.R. Greten, S.I. Grivennikov, Immunity 51 (2019) 27-41. DOI:10.1016/j.immuni.2019.06.025 |
| [29] |
S.S. Wei, J. Chi, M.M. Zhou, et al., Ind. Crop. Prod. 137 (2019) 367-376. DOI:10.1016/j.indcrop.2019.05.041 |