 2021, Vol. 32
2021, Vol. 32 


b Department of Chemistry and Biochemistry, University of South Carolina, Columbia, SC 29208, United States
Cytochromes P450 (P450s) are a large family of heme-containing redox enzymes, playing important role in the synthesis of many molecules such as steroid hormones, cholesterol and other fats and acids [1-4]. Among them, OleT, a new cytochrome P450s enzyme firstly reported by Rude and coworkers in 2011 [5], has attracted great attentions because of its ability to utilize hydrogen peroxide (H2O2) as cofactor to catalyze the decarboxylation and hydroxylation (mainly at β-position) chemistries on abundant and bioavailable fatty acids [6, 7] to generate valuable chemicals. As shown in Fig. 1, both reactions are initiated from the abstraction of a substrate hydrogen atom from the β-position of the free fatty acids by the high-valent iron (Ⅳ)-oxo heme π-cation radical intermediate (Compound I) [8, 9], followed by a competition between the OH rebound and carbon-carbon scission that deliver fatty alcohols and terminal olefin products [10].

|
Download:
|
| Fig. 1. The reaction and proposed mechanism of P450 OleT catalysis. | |
Many studies have reported that, with different substrates ( e.g., different chain lengths of fatty acids), the product profile including the ratio of terminal alkene and fatty alcohols produced by OleT catalysis was different. For eicosanoic acid, terminal alkene was identified as the sole product; while for lauric acid, more than half of the products were identified as fatty alcohols [11, 12]. However, the specific determinants that regulate the hydroxylation and decarboxylation have not been clearly elucidated so far. Researchers strived to explore the bifurcative mechanism mainly through enzyme engineering. For example, Reetz and coworkers reported some OleT mutants (Phe294Ala, Phe79Leu) could induce some trans/ cis-selectivity change on the aromatic carboxylic acids [13]. Bai et al. constructed OleT-BM3R by genetic fusion and found that the ratio of hydroxylation/decarboxylation products was dependent on chain length [14]. Makris group adopted small molecules ( e.g., norcarane) to trigger the bifurcative pathway [15]. Moreover, they found that the distal F–G loop, a structurally disordered region, was critical for mediating the product release as well as the shift of decarboxylation/hydroxylation reactions [11].
In this works, a class of structurally diversified carboxylic acids with different end groups and anchors were synthesized and analyzed as OleT non-natural substrates aiming to better understand OleT catalyzed decarboxylation and hydroxylation reaction.
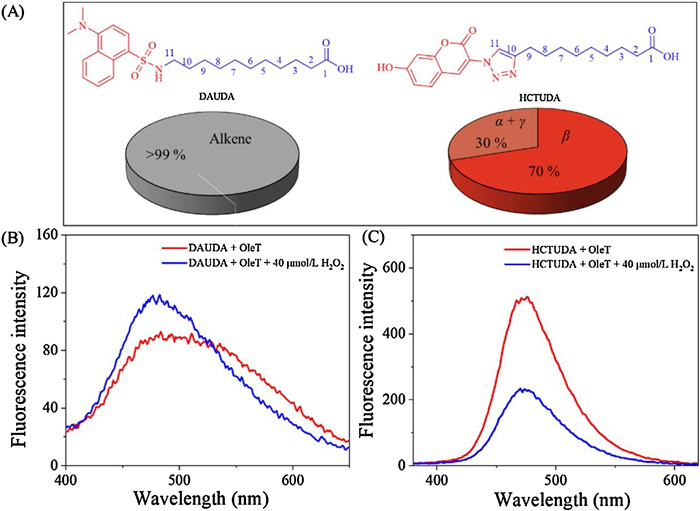
First, 11-(7-hydroxycoumarintriazolo)undecanoic acid (HCTUDA) with a coumarin anchored by a triazole in the end of fatty acid (Scheme S1 in Supporting information) and 11-(dansylamino)undecanoic acid (DAUDA) were employed to study the OleTSA based enzymatic catalysis (Fig. 1). Both compounds could be accepted as substrates of OleT despite of the bulky group attached to the fatty acid, and more than 99% of both probes were readily metabolized. More interestingly, significant different product profiles were observed. As shown in Fig. 2A, DAUDA was converted to terminal alkene as a sole product via the decarboxylation reaction (Fig. S1 in Supporting information), while HCTUDA was hydroxylated predominantly with different alcohols produced (Fig. S2 in Supporting information). These results indicate that the changes in the remote group of the substrates have profound effects on the enzyme catalyzed product distribution.

|
Download:
|
| Fig. 2. (A) Molecular structures of DAUDA and HCTUDA and their metabolic profiles (For HCTUDA, alcohol products were > 95%; Fluorescence quenching experiments of (B) DAUDA and (C) HCTUDA (OleTSA 5 μmol/L, DAUDA 35 μmol/L, HCTUDA 2 μmol/L). | |
Fluorescent probes are useful to track enzymatic reactions without the need of additional protein labeling [16]. As shown in Fig. 2B, increasing fluorescence intensity with a blue shift (17 nm) was observed for DAUDA with the addition of OleT and H2O2, which is consistent with the previous study [11]. By contrast, the fluorescence of HCTUDA was significantly quenched during the enzymatic reaction(Fig. 2C), while the enzyme only and H2O2 only had no such effect (Fig. S12 in Supporting information).
Next, more non-natural substrates containing different end groups (phenyl, tolyl and naphthyl) and anchors (triazole, sulfonyl, urea, thiourea) derivatized fatty acids were designed and synthesized to further investigate the relationship between the substrate structure and the product profile of OleT (Table 1, Figs. S3-S9 in Supporting information). We found that, with the phenyl end group and different anchors (1c-1f), all probes could be metabolized by OleT with the conversion rates of 42.1%–89.2%. Notably, the product patterns of these substrates were dramatically different. For the molecule 1c, the ratio of hydroxylated and decarboxylated products were 3:7, whereas substrate 1d mainly went hydroxylation with the only minor decarboxylated product (with 14:1 ratio). Probes 1e and 1f bearing urea and thiourea moieties exhibited similar products pattern. Interestingly, 1g and 1h with the addition of methyl group in tails showed different alcohol/alkene product ratios compared to 1c and 1d without methyl group, indicating the importance of the substrates' remote end in OleT catalysis. The enzyme showed much low activity when the naphthyl group was attached in the fatty acid tails (1i-1j).
|
|
Table 1 Metabolic profile of OleTSA with differnt fatty acid derivatives. |

{kind=link}
{kind=link}
It is well known that molecular size is important for enzymatic metabolism, as most physicochemical properties are strongly size-related [16-18], which can be estimated from 3D molecular geometries using van der Waals radius [19]. Therefore, the structures of DAUDA and HCTUDA were optimized by density-functional theory (DFT) to estimate the molecular parameters with the consideration of solvent effect. The result was shown in Table S1 (Supporting information). The structures of these two molecules are significantly different. DAUDA molecule displays twisted conformation providing a more concentrated molecule, while HCTUDA molecule stretches out displaying longer conformation. While for eicosanoic acid and lauric acid that also have dramatically different product profile, no such kind of conformational alterations were observed (Table S1). Other geometry variations ( e.g., height, shape and angle) were also observed among different fatty acid derivatives (Tables S1 and S2 in Supporting information), revealing no significant correlations based on the DFT calculation. We therefore speculate that the substrate binding pocket of OleT provides unique environment where the non-natural substrates' geometry and conformation are stringently confined, which triggers the bifurcative pathway.
To further understand the bifurcative reaction for different probes, additional molecular docking studies were carried out. The structure of OleTJE, a functional ortholog from Jeotgalicoccus sp. 8456 has been reported (PDB code of ligand free enzyme: 4L54; PDB code of eicosanoic acid bound enzyme: 4L40) [7, 20]. It shows high amino acid sequence identity (69%), and all secondary elements are identity to OleTSA [21, 22]. The ligand free OleTJE was used as template for docking [23]. OleT has an elongated hydrophobic fatty acid binding pocket and a flexible F–G loop which regulates its catalysis [7, 11]. As shown in Figs. 3A and B, the eicosanoid acid's orientation and geometry are maintained through hydrogen bonding with Arg245 and a series of hydrophobic contacts including Leu41, Phe46, Phe294, Leu176 lining the entrance and Phe173, Pro296, Pro293, Val292, Phe291 locating at the pocket. Though there is no clear evidence of His85 in the catalysis, collective studies indicate that this residual, as a potential proton donor to reactive iron-oxo species, may play significant roles during substrate decarboxylation [20, 24, 25].

|
Download:
|
| Fig. 3. (A, B) Crystal structure of eicosanoic acid-bound OleT (PDB: 4L40). (C, D) Proposed model of DAUDA bound OleT. (E, F) Proposed model of HCUDA bound OleT. The substrates were shown in green. The hydrophobic residues along the substrate binding pocket and Arg245, His58 were shown in sticks. F-G loop was highlighted in cyan. The heme was shown in red and the iron was in yellow sphere. The distance between carboxylate and Arg245 and His85 was labled. | |
{kind=link}
The predicted DAUDA bound OleT model reveals that the end group of DAUDA is sandwiched by Phe46 and Phe294, and the contacts with the hydrophobic residues are largely maintained. The position of carboxylate group of DAUDA is similar to that of eicosanoic acid. Hydrogen bonding with Arg245 is the only polar contacts (Figs. 3C and D). The distance between His85 and the carboxylate of DAUDA is 5.1Å. For comparison, a docking was done with the probe 1d which has fatty alcohols as main products (Fig. S10 in Supporting information). The predicated model shows that there is a slight shift of the carboxylate group of 1d relative to that of DAUDA, implying that the interactions between the bulky end group of the probe and the enzyme induce the position change of the carboxylate, thus affect the reaction bifurcation dramatically.
HCTUDA bond OleT model reveals that the end group of this probe slides towards to the pocket, which can be attributed to the strong polarity of the hydroxylated coumarin and the triazole anchor (Fig. 3E). Interestingly, a significant shift of the carboxylates in the active site is induced with this structure of end group (Fig. 3F). The distance between the carboxylate group and His85 is 3.1Å within hydrogen bonding distance. This position of carboxylate is more like that of lauric acid(Fig. S11 in Supporting information). We speculate that the hydrogen bonding between His85 and the carboxylates of the probes may interrupt its native function, thus lead to substrate hydroxylation. Notably, compared to the short chain of fatty acid ( i.e., lauric acid), the bulky end group of HCTUDA is more closed to the F–G loop, which may be beneficial to stabilize this unique conformation. Consequently, HCTUDA predominantly goes to the hydroxylation pathway.
In summary, a series of novel fatty acid derivatives were designed and synthesized to probe the hydroxylation/decarboxylation activity of P450 OleT. It demonstrated that most of the derivatives were well accepted as substrates of OleT, which extraordinarily broadened the enzyme's substrate scope. More importantly, diversified product profiles were observed based on the difference in end groups, which indicated that the minor change in the remote site of substrates could regulate the hydroxylation/decarboxylation reaction. Evidently the molecular docking study implies that the position shifts of the carboxylate group induced by the remote structure contributes the bifurcative catalysis. This work sheds light on the understanding of the OleT catalysis and will benefit the application of OleT enzymes in synthesis and biofuel industry [12].
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe wish to acknowledge the support from the State Scholarship Fund of the China Scholarship Council (No. 201806310084). We also wish to thank Dr. Thomas M. Makris for providing the OleTSA plasmid, thank Olivia Manley, Suman Das for their help on protein purification. We also acknowledge technical supports from the staffs at the Mass Spectrometry at the University of South Carolina.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.042.
| [1] |
I.G. Denisov, T.M. Makris, S.G. Sligar, I. Schlichting, Chem. Rev. 105 (2005) 2253-2277. DOI:10.1021/cr0307143 |
| [2] |
F.P. Guengerich, FASEB J. 6 (1992) 745-748. DOI:10.1096/fasebj.6.2.1537465 |
| [3] |
P. Anzenbacher, E. Anzenbacherová, Cell. Mol. Life Sci. 58 (2001) 737-747. DOI:10.1007/PL00000897 |
| [4] |
D.C. Lamb, M.R. Waterman, S.L. Kelly, F.P. Guengerich, Curr. Opin. Biotech. 18 (2007) 504-512. DOI:10.1016/j.copbio.2007.09.010 |
| [5] |
M.A. Rude, T.S. Baron, S. Brubaker, et al., Appl. Environ. Microb. 77 (2011) 1718-1727. DOI:10.1128/AEM.02580-10 |
| [6] |
J.L. Grant, C.H. Hsieh, T.M. Makris, J. Am. Chem. Soc. 137 (2015) 4940-4943. DOI:10.1021/jacs.5b01965 |
| [7] |
J. Belcher, K.J. McLean, S. Matthews, et al., J. Biol. Chem. 289 (2014) 6535-6550. DOI:10.1074/jbc.M113.527325 |
| [8] |
A. Dennig, M. Kuhn, S. Tassoti, et al., Angew. Chem. Int. Ed. 54 (2015) 8819-8822. DOI:10.1002/anie.201502925 |
| [9] |
A. Greule, J.E. Stok, J.J. De Voss, M.J. Cryle, Nat. Prod. Rep. 35 (2018) 757-791. DOI:10.1039/C7NP00063D |
| [10] |
J.T. Groves, G.A. McClusky, R.E. White, M.J. Coon, Biochem. Biophys. Res. Commun. 81 (1978) 154-160. DOI:10.1016/0006-291X(78)91643-1 |
| [11] |
J.A. Amaya, C.D. Rutland, N. Leschinsky, T.M. Makris, Biochemistry 57 (2018) 344-353. DOI:10.1021/acs.biochem.7b01065 |
| [12] |
L. Zhang, O.M. Manley, D. Ma, et al., Bioresour. Technol 311 (2020) 123538. DOI:10.1016/j.biortech.2020.123538 |
| [13] |
J. Wang, R. Lonsdale, M.T. Reetz, Chem. Commun. 52 (2016) 8131-8133. DOI:10.1039/C6CC04345C |
| [14] |
L. Chen, F. Shen, S. Wang, et al., ACS Catal. 8 (2018) 5794-5798. DOI:10.1021/acscatal.8b01313 |
| [15] |
C.H. Hsieh, X. Huang, J.A. Amaya, et al., Biochemistry 56 (2017) 3347-3357. DOI:10.1021/acs.biochem.7b00338 |
| [16] |
P. Buchwald, Metab Drug, Dispos. 42 (2014) 1785-1790. DOI:10.1124/dmd.114.059717 |
| [17] |
J.M. Cyphert, C.S. Trempus, S. Garantziotis, Int. J. Cell Biol. 2015 (2015) 563818. |
| [18] |
P. Buchwald, N. Bodor, J. Med. Chem. 42 (1999) 5160-5168. DOI:10.1021/jm990145k |
| [19] |
K.J. Laidler, J.H. Meiser, Physical Chemistry. 2nd ed.. Boston: Houghton Mifflin, 1995.
|
| [20] |
S. Matthews, J.D. Belcher, K.L. Tee, et al., J. Biol. Chem. 292 (2017) 5128-5143. DOI:10.1074/jbc.M116.762336 |
| [21] |
Y. Jiang, Z. Li, C. Wang, et al., Biotechnol. Biofuels 12 (2019) 79-92. DOI:10.1186/s13068-019-1419-6 |
| [22] |
J.A. Amaya, Mechanisms of Decarboxylation in the CYP152 Family of Cytochrome P450s (Dissertation), University of South Carolina, 2018.
|
| [23] |
O. Trott, A.J. Olson, J. Comput. Chem. 31 (2010) 455-461. |
| [24] |
J.L. Grant, M.E. Mitchell, T.M. Makris, PNAS 113 (2016) 10049-10054. DOI:10.1073/pnas.1606294113 |
| [25] |
M.A. Rude, T.S. Baron, S. Brubaker, Appl. Environ. Microbiol. 77 (2011) 1718-1727. DOI:10.1128/AEM.02580-10 |