 2021, Vol. 32
2021, Vol. 32 


b College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, China
Fluorinated arenes, especially difluoroalkylated arenes, remain privileged moieties for drug discovery and development owing to gem-difluoromethylene group's unique stability, and isosteric properties as an ethereal oxygen atom or a carbonyl group, as well as a lipophilic hydrogen-bond donor [1]. Accordingly, various approaches to direct difluoroalkylation of aromatic rings have been explored extensively in the last few decades [2]. Although heteroarenes can afford site-selective products due to their intrinsic electronic effects and photo-redox-catalyzed ortho-difluoroalkylation of anilines have been achieved [3], general aromatic compounds usually suffer from poor site selectivity for remote C-H difluoroalkylation. Until recently, meta-selective C-H difluoroalkylation of 2-arylpyridine derivatives [4] and purine [5] has successfully been achieved by groups of Ackermann and Wang. Following these remote C-H difluoroalkylations, para-selective counterparts have been demonstrated viable to several aromatic rings, such as aniline derivatives [6], oximes [7], ketones [8], aldehydes [9] and benzoate derivatives [10] (Scheme 1). However, given that the formation of cycloruthenation between oxygen-containing directing group and ruthenium is sluggish owing to the weak coordination of oxygen and ruthenium, the direct para-selective difluoroalkylation of aromatic aldehydes, ketones or benzoate derivatives employing less expensive ruthenium catalysts still remains unsolved and challenging [11].

|
Download:
|
| Scheme 1. The remote CH difluoroalkylation of arenes. | |
Aldehydes as the directing group (DG) in the C-H activation normally suffer from their weak coordinating ability, susceptibility toward oxidation, and undesired metal insertion into acyl C-H bond. To overcome these limitations, Yu and others recently introduced the transient directing group (TDG) concept and successfully achieved diverse ortho-C–H functionalization of aromatic aldehydes using amines as TDGs [12]. We envisioned that introduction of TDG in para-difluoroalkylation of aromatic aldehydes would enable the formation of strong coordinating imine moiety which can benefit substrate-ruthenium coordination. Herein, we report the first transient directing group promoted para-difluoroalkylation of aromatic aldehydes. This protocol employs less expensive ruthenium catalysts and allows the rapid access to both difluoroalkylated aromatic aldehydes and ketones.
We commenced our initial investigation by the reaction of benzaldehyde 1a and bromodifluoroacetate 2a using [Ru( p-cymene)Cl2]2 as catalyst with diverse TDGs (Scheme 2, Table S1 in Supporting information). When subjected to the model reactions, the reported bidentate TDGs previously employed for ortho-C–H functionalization of aldehydes afforded low yields in this remote C–H difluoroalkylation (TDG1-TDG7) [12b–e, 12k, 12l]. Aniline type monodentate TDGs did not improve the reaction efficiency (TDG8 and TDG9) [12h, 12m]. To our delight, aliphatic primary amine TDG10 could significantly enhanced the reaction outcome, delivering the para-product 3a in 68% (Table 1, entry 1). Other amines, such as 2-methylpropan-1-amine and 2-methylbutan-2-amine, gave lower chemical yields.

|
Download:
|
| Scheme 2. Transient directing group screening. Standard conditions: 1a (0.2 mmol), 2a (0.6 mmol), [Ru( p-cymene)Cl2]2 (5mol%), Na2CO3 (2 equiv.), AgTFA (2 equiv.), N-acetyl-l-isoleucine (30mol%), TDG (0.5 equiv.), DCE (1mL), 155 ℃, 36 h, under Ar. Isolated yields. | |
|
|
Table 1 Optimization of reaction conditions. |

{kind=link}
{kind=link}
Further survey of other ruthenium catalysts [7] and previous employed Pd(PPh3)4 [8] showed lower catalytic efficiency (Table 1, entries 2–6). Besides, the use of N-acetylglycine, 1-adamantane carboxylic acid (1-Ad-OH), pivalic acid ( t-BuCOOH) and 2, 4, 6-trimethylbenzoic acid (MesCOOH) instead of N-acetyl-l-isoleucine decreased the efficiency of difluoroalkylation and regioselectivity (Table 1, entries 7–10). Further screening of diverse silver salts demonstrated AgTFA as the optimal choice (Table 1, entries 11–17). A thorough investigation of bases and solvents revealed the combination of Na2CO3 and 1, 2-dichloroethane (DCE) led to the best chemical yields (Table 1, entries 18–23).
With optimal conditions, we treated different benzaldehydes with 2a to examine the functional-group tolerance (Scheme 3). Difluoroalkylation of benzaldehyde derivatives with ortho substituents proceeded smoothly to furnish the corresponding para-difluoroalkylated products 3b-3e. Pleasingly, meta-substituted benzaldehyde derivatives provided the corresponding products 3f-3i difluoroalkylated at the sterically hindered para-position in moderate to good yield. This reactivity is different to that observed in previous meta-selective alkylation and difluoroalkylation reactions [8], in which meta-substituted substrates are less reactive than their ortho- or para-substituted analogues. Notably, previous uninvestigated 2-naphthaldehyde only generated the meta-difluoroalkylated product 3j. Heteroarenes, such as furan-2-carbaldehyde and thiophene-2-carbaldehyde, are also viable substrates, providing 3k and 3l in moderate yields. The current methodology can be easily extended to the coupling of BrCF2CONMe2 (2b) and heteroaryl difluoromethyl bromide (2d) with benzaldehyde, providing 3m and 3n in good yields. Although 1H and 19F NMR of 3n is in agreement with literature [13], it was containment with aryl impurities and cannot be further purified. Unfortunately, cyclic amides, difluoromethyl halides (HCF2X), difluoroalkyl halides, BrCF2PO(OEt)2 and BrCFHCO2Et didnot react with aldehydes to provide desired products. Further extension of this protocol to aromatic ketones successfully furnished corresponding para-difluoroalkylated products as well (Scheme 3). The acetophenone derivatives performed well under the optimal conditions (4a-4d). Other alkyl aromatic ketones were compatible with the difluoroalkylation, providing the corresponding products 4e-4g in moderate to good yields. The diphenyl ketones afforded monodifluoromethylation products 4h and 4i. 9-Fluorenone, the important intermediate for organic synthesis and materials science, also provided the mono- para-difluoroalkylated product 4j. 1-Tetralone and chromanone reacted smoothly with bromodifluoroacetate to afford the corresponding para-difluoroalkylated products 4k and 4l in 56% and 53% yields, respectively. Substrates of 4m provided single difluoroalkylated products with high selectivity at the para-position of the benzoyl ring rather than the electronically rich aromatic ring. Directly introducing fluorine-containing functional groups into bioactive compounds is an effective method for new drug development. To our delight, the difluoroalkylated ketoprofen derivative (4n) and octabenzone derivative (4o) were successfully obtained by this transformation. Although 1H and 19F NMR of 4o is in agreement with literature [9a], it was containment with aryl impurities and can not be further purified.

|
Download:
|
| Scheme 3. Reaction scope. Standard conditions: 1 (0.2 mmol, 1.0 equiv.), 2 (0.6 mmol, 3.0 equiv.), [Ru( p-cymene)Cl2]2 (5mol%), Na2CO3 (0.4 mmol, 2.0 equiv.), AgTFA (0.4 mmol, 2.0 equiv.), N-acetyl-l-isoleucine (30mol%), TDG10 (0.1 mmol, 0.5 equiv.), DCE (1.0mL), 155 ℃, 36 h, under Ar; Isolated yields. aCannot be separated from aryl impurities. | |
{kind=link}
Further conversion of difluoralkylated products can also be achieved via transient directing group strategy. ortho-C–H methylation [12e], fluorination [12e], chlorination [12i] and arylation [12k] were successfully applied to functionalize 3a. In contrast to methylation, fluorination and chlorination, mono- and diarylation of 3a provided 5a and 5b with 1:1 ratio (Scheme 4).

|
Download:
|
| Scheme 4. Transformations of 3a. | |
{kind=link}
To explore the mechanism and the explanation for the para-selectivity, a series of control experiments were carried out. Addition of TEMPO as radical inhibitor completely suppressed difluoroalkylation, implying a plausible radical pathway (Scheme 5a). Trapping of difluoroalkyl radical using 1, 1-diphenylethylene under the standard conditions was detected via 19F NMR, affording a mixture of 6 and 7 (Scheme 5b) [14]. This result suggests a difluroalkyl radical is involved in the para-selective reaction. As formation of chelation-assisted cycloruthenation is normally the key factor that controls the regioselectivity of meta- or para-C–H difluroakylation, we subjected the deuterated substrate [D8]-1d for investigation of cycloruthenation. The product [D8]-3l without any D/H exchange by NMR analysis implies a distinct coordination mode from previous cycloruthenation (Scheme 5c) [7].

|
Download:
|
| Scheme 5. Preliminary mechanistic study. | |
{kind=link}
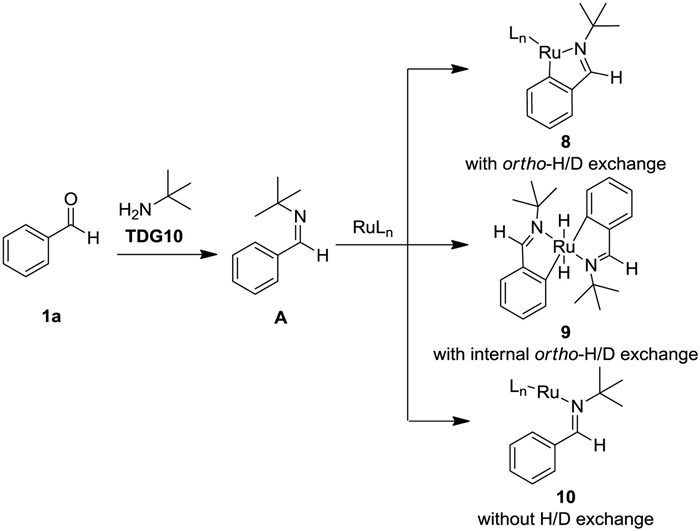
Another plausible pathway without D/H exchange would be formation of complex 9 (Scheme 6) [15]. To elucidate this possibility, cross-over H/D exchange experiment was carried out, which substrates [D8]-1d and 1e were recovered without H/D exchange (Scheme 5d). These results indicate the cycloruthenation cannot be obtained from coordination of A with Ru catalysts under our conditions and is not responsible for para-selectivity. When substrate 1a was subjected to the difluoroalkyl radical generated conditions without Ru catalysts [16], a mixture of para- and meta-difluoroalkylated products was obtained in 5% yield (Scheme 5e). Therefore, we hypothesized para-selectivity could be controlled by the steric and electronic feature of complex B, which is similar to Zhou's reports [17].

|
Download:
|
| Scheme 6. Plausible coordination mode of ruthenium complex with imine moieties. | |
{kind=link}
Based on these experiments, a mechanism is proposed for para-selective difluoroalkylation of aromatic aldehydes and ketones (Scheme 7). First, the TDG10 reacts with the 1a to form imine intermediate A. Subsequent coordination of A and F affords complex B. A radical C, derived from 2-bromo-2, 2-difluoroacetate via a single-electron-transfer process [7], is trapped by B to generate D, which releases product 3a, TDG10 and catalysis specices F.

|
Download:
|
| Scheme 7. Proposed catalytic cycle. | |
{kind=link}
In summary, we have developed a general transient directing group strategy for para-selective C-H difluoroalkylation of aromatic aldehydes and ketones. The protocol can be performed by using an inexpensive ruthenium catalyst, and allows the rapid access to challenging para-difluoroalkylated aldehydes. Mechanism investigation suggests that the distinct coordination of ruthenium complex with imine moieties is responsible for para-selectivity.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThe authors gratefully acknowledge support from the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (No. UNPYSCT-2017124). Dr. Guijie Mao was acknowledged for her help in the NMR spectrum.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.044.
| [1] |
(a) G.M. Blackburn, D.A. England, F. Kolkmann, J. Chem. Soc. Chem. Commun. (1981) 930-932; (b) M.O. Anderson, J. Zhang, Y. Liu, et al., J. Med. Chem. 55 (2012) 5942-5950; (c) J.O. Link, J.G. Taylor, L. Xu, et al., J. Med. Chem. 57 (2014) 2033-2046; (d) N.A. Meanwell, J. Med. Chem. 54 (2011) 2529-2591; (e) S. Purser, P.R. Moore, S. Swallow, et al., Chem. Soc. Rev. 37 (2008) 320-330. |
| [2] |
(a) M.C. Belhomme, T. Poisson, X. Pannecoucke, J. Org. Chem. 79 (2014) 7205-7211; (b) M.C. Belhomme, A. Bayle, T. Poisson, et al., Eur. J. Org. Chem. 2015 (2015) 1719-1726; (c) C. Chen, R. Zeng, J. Zhang, et al., Eur. J. Org. Chem. 2017 (2017) 6947-6950; (d) H. Chen, P. Li, M. Wang, et al., Org. Lett. 18 (2016) 4794-4797; (e) S. Han, A. Liang, X. Ren, et al., Tetrahedron Lett. 58 (2017) 4859-4863; (f) J. Jung, E. Kim, Y. You, et al., Adv. Synth. Catal. 356 (2014) 2741-2748; (g) M.L. Ke, Q.L. Song, Chem. Commun. 53 (2017) 2222-2225; (h) J.A. Leitch, C.J. Heron, J. McKnight, et al., Chem. Commun. 53 (2017) 13039-13042; (i) J.A. Leitch, C.L. McMullin, M.F. Mahon, et al., ACS Catal. 7 (2017) 2616-2623; (j) S. Murakami, H. Ishii, T. Tajima, Tetrahedron 62 (2006) 3761-3769; (k) Y. Ohtsuka, T. Yamakawa, Tetrahedron 67 (2011) 2323-2331; (l) Y.M. Su, Y. Hou, F. Yin, et al., Org. Lett. 16 (2014) 2958-2961; (m) L. Wang, X.J. Wei, W.L. Jia, Org. Lett. 16 (2014) 5842-5845; (n) L. Wang, X.J. Wei, W.L. Lei, et al., Chem. Commun. 50 (2014) 15916-15919; (o) Z. Feng, Q.Q. Min, Y.L. Xiao, et al., Angew. Chem. Int. Ed. 53 (2014) 1669-1673; (p) Z. Feng, Y.L. Xiao, X. Zhang, Acc. Chem. Res. 51 (2018) 2264-2278; (q) Y.L. Xiao, W.H. Guo, G.Z. He, et al., Angew. Chem. Int. Ed. 53 (2014) 9909-9913. |
| [3] |
(a) L.C. Yu, J.W. Gu, S. Zhang, et al., J. Org. Chem. 82 (2017) 3943-3949; (b) P.B. Arockiam, L. Guillemard, J. Wencel-Delord, Adv. Synth. Catal. 359 (2017) 2571-2579. |
| [4] |
(a) Z. Ruan, S.K. Zhang, C. Zhu, et al., Angew. Chem. Int. Ed. 56 (2017) 2045-2049; (b) Z.Y. Li, L. Li, Q.L. Li, et al., Chem. Eur. J. 23 (2017) 3285-3290; (c) H. Zhao, G. Ma, X. Xie, et al., Chem. Commun. 55 (2019) 3927-3930; (d) P. Gandeepan, J. Koeller, K. Korvorapun, et al., Angew. Chem. Int. Ed. 58 (2019) 9820-9825. |
| [5] |
F. Fumagalli, S. Warratz, S.K. Zhang, et al., Chem. Eur. J. 24 (2018) 3984-3988. DOI:10.1002/chem.201800530 |
| [6] |
(a) X.G. Wang, Y. Li, L.L. Zhang, et al., Chem. Commun. 54 (2018) 9541-9544; (b) C. Yuan, L. Zhu, C. Chen, et al., Nat. Commun. 9 (2018) 1189. |
| [7] |
C. Yuan, L. Zhu, R. Zeng, et al., Angew. Chem. Int. Ed. 57 (2018) 1277-1281. DOI:10.1002/anie.201711221 |
| [8] |
G. Tu, C. Yuan, Y. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 15597-15601. DOI:10.1002/anie.201809788 |
| [9] |
(a) J. Zhou, F. Wang, Z. Lin, et al., Org. Lett. 22 (2020) 68-72; (b) W. Tang, F. Tang, J. Xu, et al., Chem. Commun. 56 (2020) 1497-1500. |
| [10] |
Y.J. Mao, B.X. Wang, Q.Z. Wu, et al., Chem. Commun. 55 (2019) 2019-2022. DOI:10.1039/C8CC09129C |
| [11] |
(a) X.F. Huang, Q.L. Wu, J.S. He, et al., Org. Biomol. Chem. 13 (2015) 4466-4472; (b) Z. Jiao, L.H. Lim, H. Hirao, et al., Angew. Chem. Int. Ed. 57 (2018) 6294-6298; (c) W. Li, D. Yuan, G. Wang, et al., J. Am. Chem. Soc. 141 (2019) 3187-3197; (d) K.D. Mane, A. Mukherjee, K. Vanka, et al., J. Org. Chem. 84 (2019) 2039-2047. |
| [12] |
(a) P.W. Tan, N.A.B. Juwaini, J. Seayad, J. Org, Lett. 15 (2013) 5166-5169; (b) F.L. Zhang, K. Hong, T.J. Li, et al., Science 351 (2016) 252-256; (c) X.Y. Chen, S. Ozturk, E.J. Sorensen, Org. Lett. 19 (2017) 1140-1143; (d) X.Y. Chen, S. Ozturk, E.J. Sorensen, Org. Lett. 19 (2017) 6280-6283; (e) X.Y. Chen, E.J. Sorensen, J. Am. Chem. Soc. 140 (2018) 2789-2792; (f) X.Y. Chen, E.J. Sorensen, Chem. Sci. 9 (2018) 8951-8956; (g) A.E. Hande, V.B. Ramesh, K.R. Prabhu, Chem. Commun. 54 (2018) 12113-12116; (h) F. Li, Y. Zhou, H. Yang, et al., Org. Lett. 20 (2018) 146-149; (i) F. Li, Y. Zhou, H. Yang, et al., Org. Lett. 21 (2019) 3692-3695; (j) X. Liu, Z. Wang, Q. Chen, et al., Appl. Organomet. Chem. 32 (2018) e4039; (k) X.H. Liu, H. Park, J.H. Hu, et al., J. Am. Chem. Soc. 139 (2017) 888-896; (l) F. Ma, M. Lei, L. Hu, Org. Lett. 18 (2016) 2708-2711; (m) D. Mu, X. Wang, G. Chen, et al., J. Org. Chem. 82 (2017) 4497-4503; (n) D.Y. Wang, S.H. Guo, G.F. Pan, et al., Org. Lett. 20 (2018) 1794-1797; (o) X. Wang, S. Song, N. Jiao, Chin. J. Chem. 36 (2018) 213-216; (p) Y. Cheng, J. Zheng, C. Tian, et al., Asian J. Org. Chem. 8 (2019) 526-531. |
| [13] |
Y.L. Xiao, B. Zhang, Z. Feng, et al., Org. Lett. 16 (2014) 4822-4825. DOI:10.1021/ol502121m |
| [14] |
Q. Wang, Y.T. He, J.H. Zhao, et al., Org. Lett. 18 (2016) 2664-2667. DOI:10.1021/acs.orglett.6b01038 |
| [15] |
Q. Yu, L. Hu, Y. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 15284-15288. DOI:10.1002/anie.201507100 |
| [16] |
I.S. Kondratov, M.Y. Bugera, N.A. Tolmachova, et al., J. Org. Chem. 80 (2015) 12258-12264. DOI:10.1021/acs.joc.5b02171 |
| [17] |
Z. Jiao, L.H. Lim, H. Hirao, et al., Angew. Chem. Int. Ed. 57 (2018) 6294-6298. |