 2021, Vol. 32
2021, Vol. 32 


b State Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Lanzhou 730000, China;
c The Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, Shihezi University, Shihezi 832004, China
Chemodivergent reaction to selectively construct diverse compounds from readily available starting materials has proven to be one of the most important strategies in organic synthesis [1]. It attracts more and more attention and the power to develop chemodivergent system is a longstanding challenging and an important task for organic chemists. This strategy has been used not only to develop eco-efficient route to access novel biologically active small molecules [2], but also for creating molecular complexity to facilitate drug discovery [3]. Many effective reaction models, such as condition-controlled [4, 5], catalyst-mediated [6, 7], additive-controlled [8, 9], directing group-enabled [10] and intermediate-dependent strategy [11, 12] were all involved, and with which, a variety of molecular frameworks were synthesized in modifying either chemo-, regio-, or stereoselectivity [13-15].
Acetals are important synthetic intermediates that are generally used as carbonyl protection in multistep synthesis reactions [16, 17]. 2, 2-Dimethoxyacetaldehyde is a cheap and readily available biomass-derived platform molecule, because it can be synthesized from ethylene glycol or glycerol [18, 19]. This acetal is known to serve as a building block for the synthesis of a structurally diverse set of molecules. Recently, intense research efforts have been dedicated to developing conversion pathway of valuable biomass platform molecules [20]. In literature, 2, 2-dimethoxyacetaldehyde was, simply used as a two-carbon electrophilic regent in most of the cases. For instance, 2, 2-dimethoxyacetaldehyde reacted readily with amine or methylene compound (Scheme 1, path a [21]). In most of the cases, only the aldehyde carbonyl participated in the reaction, and the dimethoxymethylene fragment remained unaffected [22]. In some special cases, 2, 2-dimethoxyacetaldehyde contributed two of its carbons to form some heterocyclic compounds (Scheme 1, paths c and d) [23]. Although these individual examples demonstrated that the two electrophilic carbons of 2, 2-dimethoxyacetaldehyde are both reactive, the potential of this reagent in diversity-originated synthesis has not fully displayed, particularly for creating the molecular complexity starting from the same substrates.

|
Download:
|
| Scheme 1. Reactions of 2, 2-dimethoxyacetaldehyde. | |
Direct C-C bond cleavage has attracted much attention because of the great ability to simplify the synthetic procedure [24]. Removal of carbonyl group via oxidative cleavage is one of the typical ways [25-27], which strategically uses a carbonyl group as a temporary activating or directing group in a number of synthetic processes. Chi and co-workers [28] reported an interesting approach, in which the C-C bond cleavage of chiral aldehydes occurred through an in situ generated enamine intermediate, and the following oxidative cleavage of C-C bond by oxygen. Based on this, we envisioned that the reaction of 2, 2-dimethoxyacetaldehyde and aniline might deliver phenylformamide (Scheme 1, bottom). On the other hand, the 1, 2-enaminol generated from α-hydroxy ketones/alderhyde and aniline through Heyns rearrangement have been widely used to synthesize α-amino carbonyl compounds [29-31]. On the basis of this unique rearrangement mechanism, methyl phenylglycinate should also be hopefully formed from 2, 2-dimethoxyacetaldehyde and aniline (Scheme 1, bottom). In addition, because of the fact that Heyns rearrangement occurs generally under acidic conditions, downstream conversion of the generated methyl phenylglycinate is also possible. This may allowed us to develop some new reactions that are not possible with the previous reported alkaline conditions or metal carbene catalysts [32, 33]. Out of all these considerations, we started to use 2, 2-dimethoxyacetaldehyde as a starting reagent to explore diversified reactions for organic synthesis. Herein, we reported the successful outcome of our endeavor, in which three products including N-phenylformamide, methyl phenylglycinate and dimethyl 2, 2′-(phenylazanediyl)diacetate, were synthesized in good yield from the same substrates, 2, 2-dimethoxyacetaldehyde and aniline, and in these reactions, the product distribution can be finely tuned by changing the reaction parameters.
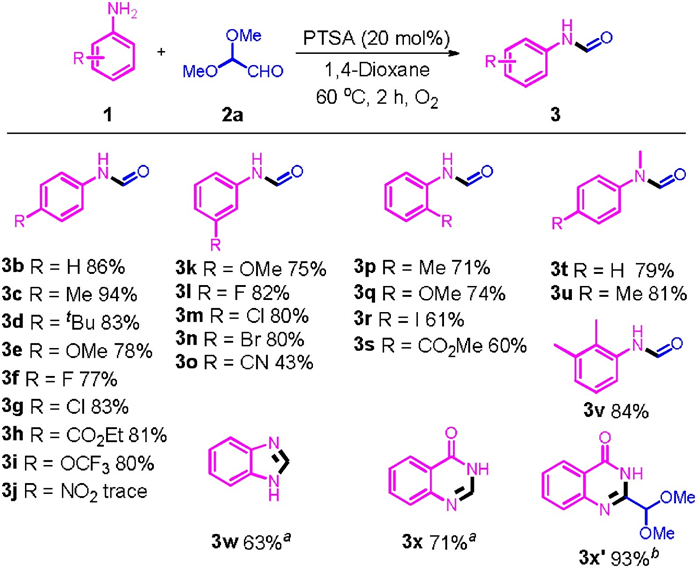
To validate our hypothesis, we started our study by using 4-bromoaniline (1a) and 2, 2-dimethoxyacetaldehyde (2a) as model substrates. Initially, the reaction was performed in 1, 4-dioxane under air. No reaction occurred in the absence of catalyst (Table 1, entry 1). When strong Lewis acids, Sc(OTf)3 and AlCl3, were used as catalysts, it is difficult to initiate the reaction neither (entries 2 and 3). When a stoichiometric amount of a weak Lewis acid, MnCl2, was added, N-(4-bromophenyl)formamide 3a was formed exclusively, however, the yield reached only 23% after 2 h of reaction at 80 ℃ (entry 4). The yield of 3a can be improved to 42% by using Fe(OTf)3 as a catalyst (entry 5). PTSA exhibited excellent catalytic activity for this reaction, with which 3a can be obtained in 65% yield (entry 6). Subsequently, the effect of solvent was investigated by using PTSA as catalyst. Only trace amount of 3a was detected when toluene was used as solvent (entry 7). The reaction proceeded hardly in acetonitrile and nitromethane (entries 8 and 9). When DMSO was employed as solvent, 1a was converted quickly. However, three products, including 3a, methyl phenylglycinate (4aa) and dimethyl 2, 2′-(phenylazanediyl)diacetate (4ba), were isolated with a product distribution in favor of the latter two (entry 10). The reaction proceeded also smoothly in chloroform with 4ba been formed as the major product, and after 2h of reaction, its yield reached 40% along with simultaneous isolation of 19% of 4aa and 5% of 3a (entry 11). We next changed many other parameters in order to maximize the yield of each reaction pathway. The uses of an excess amount of 1a and a protection with oxygen balloon were proved to be helpful for improving the selectivity to 3a (entries 12–14). These observations guided us to find conditions A to synthesize 3a, that are PTSA catalyst, 1, 4-dioxane solvent, a 2.0/1.0 ratio of 1a/ 2a, protection with O2 balloon, 2h and 60 ℃. Under these conditions, 3a can be isolated in 84% yield (entry 14). Somehow, performing the reaction at a higher temperature, 80 ℃, imposed a negative effect on the reaction (entry 15). The reaction selectivity was quite sensitive to the reaction time. When it was increased to 8h, the yield of 3a decreased to 32%, and the N-carboxyethylation product of aniline, 4aa, can be isolated with 38% yield (entry 16). Intriguingly, by decreasing the ratio of 1a/ 2a, the reaction selectivity can also be tuned to favoring the formation of 4aa. A 74% of yield can be obtained after 8h in a reaction with equal amount of 1a and 2a under open-air conditions (entry 17). All these observations led us to find conditions B to implement the N-carboxyethylation of 1a, that are PTSA catalyst, 1, 4-dioxane solvent, a 1.0/1.2 ratio of 1a/ 2a, protection with nitrogen balloon, 10h and 80 ℃. Under these conditions, 4aa can be isolated in 82% yield (entry 20). It should be noted that, in this case, the yield was calculated with respect to the component with lower loading, 1a. Inspired by the observation in performing the reaction in chloroform solvent (entry 11), conditions C for producing 4ba were also identified, which were confirmed to be the followings: PTSA catalyst, chloroform solvent, a 1.0/2.0 ratio of 1a/ 2a, protection with nitrogen balloon, 60 ℃, and 8h (Table S1 in Supporting information for detailed optimization studies). These conditions enabled us to synthesize 4ba in 79% yield (entry 21). In conclusion, N-(4-bromophenyl)formylation (3a) was synthesized under condition A: 1a (0.6 mmol), 2a (0.3 mmol) and 20 mol% of PTSA were added in 1, 4-dioxane at 60 ℃ for 2h under O2. Methyl phenylglycinate (4aa) was obtained under condition B: 1a (0.3 mmol), 2a (0.36 mmol) and 20 mol% of PTSA were added in 1, 4-dioxane at 80 ℃ for 10h under N2. 2, 2′-(Phenylazanediyl)diacetates (4ba) was synthesized under condition C: 1a (0.3 mmol), 2a (0.6 mmol) and 20 mol% of PTSA were added in CHCl3 at 60 ℃ for 8h under N2. Having determined the optimized conditions A, B and C, the scope of anilines bearing various functional groups for synthesis of N-phenylformamides, N-carboxyethylation and 2, 2′-(phenylazanediyl)diacetates was then explored and the results are summarized in Scheme 2, Scheme 3. Upon repeating the reactions of 2a and anilines with different functional groups under the conditions A, N-phenylformamides 3b– 3v can be synthesized with yields ranging from 43% to 94%. para-Substituted anilines with a functional group, like methyl (3c), tert-butyl (3d), methoxy (3e), halogen (3f and 3g), ethoxycarbonyl (3h) and trifluoromethyl (3i), are all tolerated the condition A. However, 4-nitroaniline can hardly be used as substrate, and the expected product, 3k, was formed only in a trace amount. meta-Substituted anilines are also readily participated in the N-formylation reaction, giving the expected products 3k– 3o in the yields ranging from 43% to 82%. More sterically demanding ortho-substituted anilines also engaged successfully in the N-formylation reaction. Under the conditions A, the reactions of N-methyl-substituted anilines proceeded also very well, and the expected product, 3t and 3u, were isolated in 78% and 81% yields, respectively. By means of a wise use of an ortho-substituted aniline, some heterocyclic compounds can be synthesized with slight modification of the condition A. For example, o-phenylenediamine can be converted to benzimidazole 3w in 63% of yield with the use of 3.0 equiv. of tert-butyl hydroperoxide (TBHP) as an oxidizing reagent under the conditions A. Anthranilamide can be converted to 4-hydroxyquinazoline 3x in 73% yield under the similar conditions. Literature survey stated that a precedent of transforming anthranilamide to 3y was established based on C-C bond cleavage, however, it involves the use of an expensive reagent, alkynyl ketone, as substrate [34]. The combination of 2a and TPHP allowed this transformation to be realized by a cost-effective way. Interestingly, a congener of 3x without C-C bond cleavage, 3x', can be obtained in 93% yield under N2 atmosphere in chloroform solvent. The presence of an acetal fragment adjacent to the nucleophilic aromatic ring may facilitate the downstream conversion of this unique compound.
|
|
Table 1 Optimization of conditions for the reaction 4-bromoaniline and 2, 2-dimethoxyacetaldehyde.a |

{kind=link}

|
Download:
|
| Scheme 2. Substrate scope of PTSA-catalyzed N-formylation reactions of anilines with 2a: 1a (0.60 mmol), 2a (0.30 mmol), PTSA (20 mol%), 1, 4-dioxane (1.0 mL), under O2 atmosphere, 60 ℃, 2 h, isolated yield, calculated with respect to 2a (reaction conditions A). TBHP (3 equiv.) as oxidant. 1a (0.30 mmol), 2a (0.30 mmol), PTSA (20 mol%), CHCl3 (1.0 mL), under N2 atmosphere, 60 ℃, 2 h. | |
{kind=link}

|
Download:
|
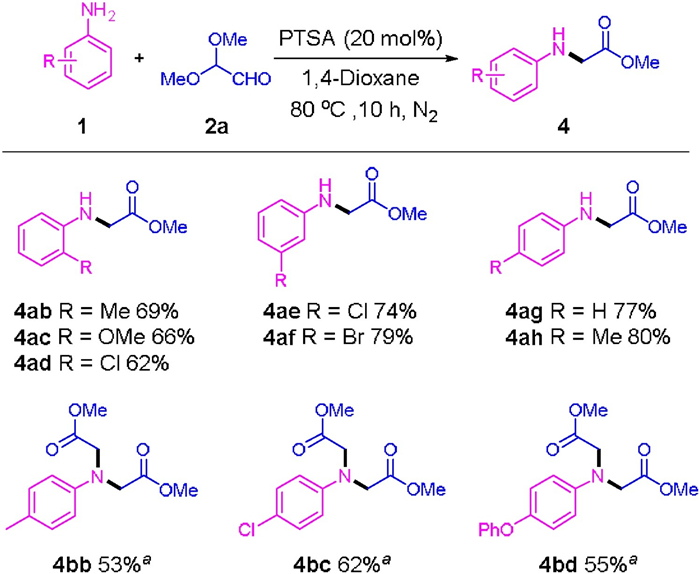
| Scheme 3. Synthesis of methyl phenylglycinate. 1 (0.3 mmol), 2a (0.36 mmol), PTSA (20 mol%), 1.4-dioxane (1.0 mL), under N2 at 80 ℃ for 10 h. Isolated yield, calculated with respect to the aniline component (reaction condition B). 1 (0.3.0 mmol), 2a (0.60 mmol), PTSA (20 mol%), CHCl3 (1.5 mL), 60 ℃, under N2 for 8 h, isolated yield, calculated with respect to the aniline (reaction condition C). | |
{kind=link}
The substrates scope of the N-carboxyethylation under the condition B is shown in Scheme 3. Anilines bearing electron-withdrawing, electron-donating, and halogen groups at the different positions all reacted smoothly with 2a, affording the N-carboxyethylation products in moderate to good yields. The reaction could well tolerate methyl, methoxyl and halogens groups, giving 4ab to 4ah in 69%–80%. It should be emphasized that, electronic effect had no obvious influence on the N-carboxyethylation reaction. ortho-Substituted anilines were found to be less reactive than those para-or meta-substituted counterparts. The scope of the double N-dicarboxyethylation reactions of anilines under the condition C is also examined. As shown in Scheme 3, the examined para-substituted anilines were all compatible with this transformation, giving direct access to the corresponding 2, 2′-(phenylazanediyl)diacetates (4bb– 4bd) in moderate yields (53%−62%).
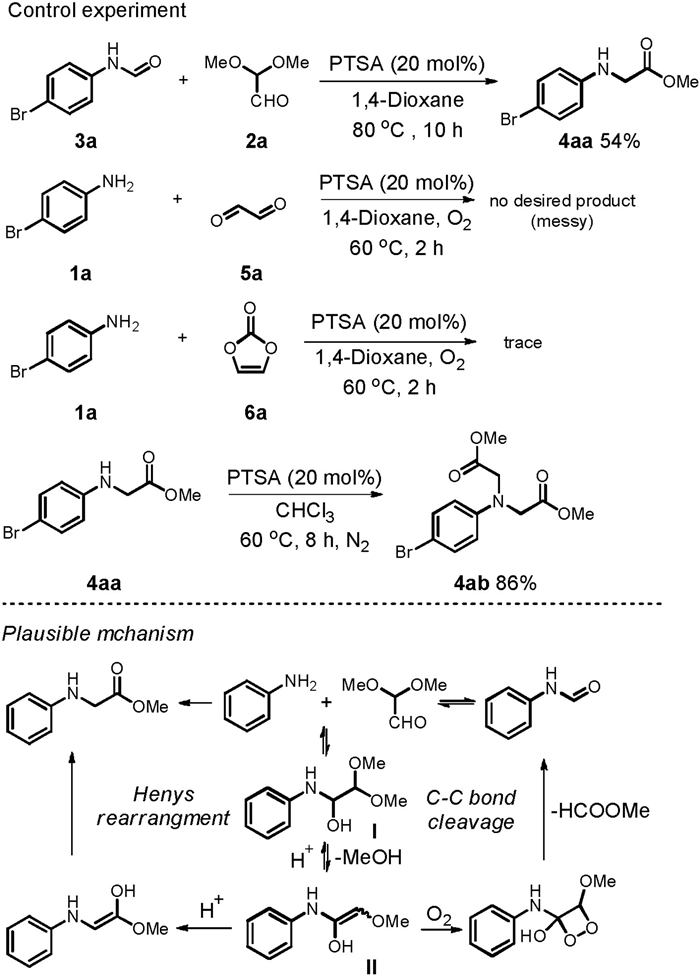
To gain the inside information of the reactions, some comparative experiments were conducted, and the result are given in Scheme 4. Treatment of 3a with 2a in 1, 4-dioxane at 80 ℃ in the presence of PTSA led to the formation of 4aa (Scheme 4). Although only moderate yield was obtained because of the low conversion of 3a, the result in Scheme 4 indicated that the formation of 3a from 1a and 2a might be reversible [35]. Replacement of 2, 2-dimethoxyacetaldehyde with either glyoxal aqueous solution or vinylene carbonate failed to form any target product under the standard conditions (Scheme 4). With reference to literature precedent [36-38], a plausible mechanism for the reactions was proposed (Scheme 4, bottom). In the initial stage of the reaction, aniline acted as a nucleophile to attack the carbonyl group of 2a, leading to the formation of an imine intermediate Ⅰ followed by imine-enamine tautomerisation to generate itermediate Ⅱ. In the presence of molecular oxygen, a [2+2] cycloaddition occurred [28, 36-39], and the following downstream decomposition of a dioxetane intermediate Ⅲ enabled the formation of 3a. In the absence of oxygen, the intermediate Ⅱ underwent enol-ketone tautomerization to give product 4aa through intermediate Ⅳ [40]. Compound 4ab was formed through a second run of the N-carboxyethylation.

|
Download:
|
| Scheme 4. Some control experiments and plausible mechanism. | |
{kind=link}
In summary, we developed an acid-catalyzed chemodivergent method for the synthesis of N-phenylformamides and methyl phenylglycinates by using the same starting substrates, 2, 2-dimethoxyacetaldehyde and anilines. By tuning the reaction conditions, the product distribution can be finely changed, thus offering an effective way for maximizing the product diversity. This reaction will provide a good example for conditions-controlled chemodivergent conversion of biomass-derived platform molecules 2, 2-dimethoxyacetaldehyde.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe thank the National Natural Science Foundation of China for financial support (Nos. 2171101076, 21872060 and 21902054), Fundamental Research Funds for the Central Universities (No. 2019kfyXJJS072), and Natural Science Foundation of Hubei Province (No. 2019CFB219). We are grateful to the Analytical and Testing Centre of HUST.
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.033.
| [1] |
B.M. Trost, Science 219 (1983) 245-250. DOI:10.1126/science.219.4582.245 |
| [2] |
W.R.J.D. Galloway, A. Isidro-Llobet, D.R. Spring, Nat. Commun. 1 (2010) 1-13. |
| [3] |
S.L. Schreiber, Science 287 (2000) 1964-1969. DOI:10.1126/science.287.5460.1964 |
| [4] |
G.X. Zhu, W.D. Shi, H. Gao, et al., Org. Lett. 21 (2019) 4143-4147. DOI:10.1021/acs.orglett.9b01333 |
| [5] |
J.B. Peng, X.F. Wu, Angew. Chem. Int. Ed. 57 (2018) 1152-1160. DOI:10.1002/anie.201709807 |
| [6] |
S. Dhole, W.J. Chiu, C.M. Sun, Adv. Synth. Catal. 361 (2019) 2916-2925. DOI:10.1002/adsc.201900088 |
| [7] |
J.F. Xu, Y. Luo, H.P. Xu, et al., J. Org. Chem. 82 (2017) 3561-3570. DOI:10.1021/acs.joc.7b00090 |
| [8] |
P. Pham, K. Nguyen, H.B. Pham, et al., Org. Lett. 21 (2019) 8795-8799. DOI:10.1021/acs.orglett.9b03414 |
| [9] |
J.Y. Wang, P. Wu, J.L. Wu, et al., J. Org. Chem. 83 (2018) 5931-5946. DOI:10.1021/acs.joc.8b00414 |
| [10] |
J.H. Cen, Y.D. Wu, J.X. Li, et al., Org. Lett. 21 (2019) 2090-2094. DOI:10.1021/acs.orglett.9b00373 |
| [11] |
W.Y. Ai, Y.Q. Liu, Q. Liu, et al., Org. Lett. 20 (2018) 409-412. DOI:10.1021/acs.orglett.7b03707 |
| [12] |
H.H. Zhang, Z.Q. Zhu, T. Fan, et al., Adv. Synth. Catal. 358 (2016) 1259-1288. DOI:10.1002/adsc.201501063 |
| [13] |
Z.H. Qu, F. Zhang, G.J. Deng, et al., Org. Lett. 21 (2019) 8239-8243. DOI:10.1021/acs.orglett.9b02978 |
| [14] |
J.Q. Sun, D.C. Bai, P.Y. Wang, et al., Org. Lett. 21 (2019) 1789-1793. DOI:10.1021/acs.orglett.9b00363 |
| [15] |
M.B. Li, A.K. Inge, D. Posevins, et al., J. Am. Chem. Soc. 140 (2018) 14604-14608. DOI:10.1021/jacs.8b09562 |
| [16] |
G. Satori, R. Ballini, F. Bigi, et al., Chem. Rev. 104 (2004) 199-250. DOI:10.1021/cr0200769 |
| [17] |
C. Liu, Y. Shen, Z. Xiao, et al., Green Chem. 21 (2019) 4030-4034. DOI:10.1039/C9GC01554J |
| [18] |
A. Guarna, A. Trabocchi, G. Menchi, E-EROS Encyclopedia of Reagents for Organic Synthesis. Chichester, UK: John Wiley & Sons, Ltd., 2007.
|
| [19] |
G.A. Bhat, A. Rajendran, R. Murugavel, Eur. J. Inorg. Chem. 6 (2018) 795-804. |
| [20] |
J.M. Ravasco, C.M. Monteiro, F. Siopa, et al., ChemSusChem 12 (2019) 4629-4635. DOI:10.1002/cssc.201902051 |
| [21] |
F.J.N. Moles, A. Bañón-Caballero, G. Guillena, et al., Tetrahedron Asymmetry 25 (2014) 1323-1330. DOI:10.1016/j.tetasy.2014.08.012 |
| [22] |
J. Yang, X. Wang, A. El-Harairy, et al., Mol. Catal. 468 (2019) 36-43. DOI:10.1016/j.mcat.2019.02.010 |
| [23] |
M. Mottinelli, M.P. Leese, B.V.L. Potter, et al., Beilstein J. Org. Chem. 13 (2017) 1871-1878. DOI:10.3762/bjoc.13.182 |
| [24] |
R.H. Crabtree, Nature 408 (2000) 415-416. DOI:10.1038/35044164 |
| [25] |
G. Hu, K. Ramakumar, S.E. Brenner-Moyer, J. Org. Chem. 82 (2017) 6972-6977. DOI:10.1021/acs.joc.7b00784 |
| [26] |
S.A. Shipilovskikh, A.E. Rubtsov, A.V. Malkov, Org. Lett. 19 (2017) 6760-6762. DOI:10.1021/acs.orglett.7b03512 |
| [27] |
Q. Xing, H. Lv, C. Xia, et al., Chem. Commun. 52 (2016) 489-492. DOI:10.1039/C5CC07390A |
| [28] |
B. Tiwari, J. Zhang, Y.R. Chi, Angew. Chem. Int. Ed. 51 (2012) 1911-1914. DOI:10.1002/anie.201107473 |
| [29] |
K. Gallas, G. Pototschnig, F. Adanitsch, et al., Beilstein J. Org. Chem. 8 (2012) 1619-1629. DOI:10.3762/bjoc.8.185 |
| [30] |
A. Frongia, F. Secci, F. Capitta, et al., Chem. Commun. 49 (2013) 8812-8814. DOI:10.1039/c3cc45278f |
| [31] |
G.X. Li, L. Tang, H.X. Liu, et al., Org. Lett. 18 (2016) 4526-4529. DOI:10.1021/acs.orglett.6b02133 |
| [32] |
D. Azarifar, H.G. Bosra, M. Zolfigol, et al., Heterocycles 68 (2006) 175-181. DOI:10.3987/COM-05-10574 |
| [33] |
M.R. Fructos, T.R. Belderrain, P. de Fremont, et al., Angew. Chem. Int. Ed. 44 (2005) 5284-5288. DOI:10.1002/anie.200501056 |
| [34] |
X.F. Yang, G.L. Cheng, J.H. Shen, et al., Org. Chem. Front. 2 (2015) 366-368. DOI:10.1039/C4QO00260A |
| [35] |
Y.F. Wei, D.X. Shi, Z. Wei, et al., Youji Huaxue 32 (2012) 1126-1130. |
| [36] |
R.L. Merzel, A.J. Fry, J. Electro. Soc. 159 (2012) G117-G122. DOI:10.1149/2.039210jes |
| [37] |
W.H. Richardson, F.C. Montgomery, M.B. Yelvington, et al., J. Am. Chem. Soc. 96 (1974) 7525-7532. DOI:10.1021/ja00831a022 |
| [38] |
A. Asha, J. Ravindran, S. Suma, et al., ChemistrySelect 5 (2020) 2545-2550. DOI:10.1002/slct.201904948 |
| [39] |
H. Sun, C. Yang, F. Gao, et al., Org. Lett. 15 (2013) 624-627. DOI:10.1021/ol303437m |
| [40] |
A. Frongia, F. Secci, F. Capitta, et al., Chem. Commun. 49 (2013) 8812-8814. DOI:10.1039/c3cc45278f |