 2021, Vol. 32
2021, Vol. 32 


It is very important to obtain chiral alcohols which have been found widespread application in pharmaceuticals, agrochemicals, fragrances, and flavors industries. The asymmetric reduction of prochiral ketones is one of the most effective and valuable methods to attain chiral alcohols [1]. Over recent decades, asymmetric transfer hydrogenation (ATH), which uses reducing agents as a source of hydrogen, such as iPrOH or HCOONa, has emerged as an appealing method due to its easy availability of hydrogen sources and the operational simplicity [2-4].
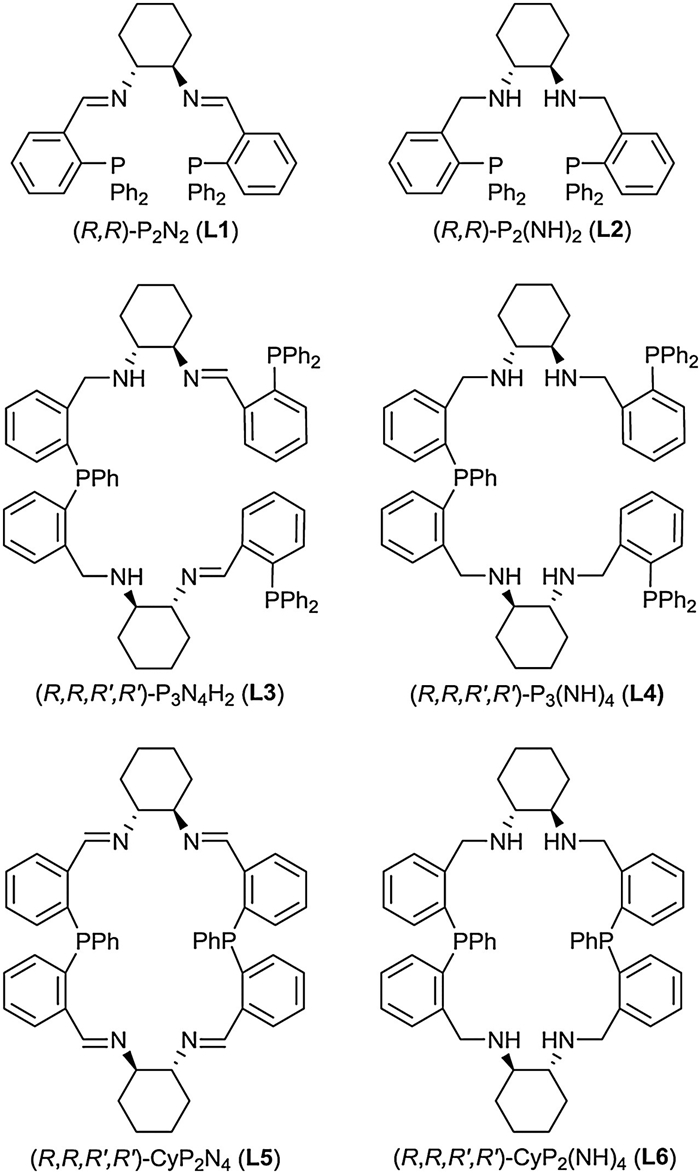
In recent years, the catalytic transformations based on non-precious metal catalyst have attracted more and more attention in both labs and industries because of the low cost, high abundance and low toxicity of these cheap metals [5]. As the third most abundant transition metal in the earth's crust, manganese thus has the potential to be used as a nontoxic and readily available alternative to noble metals in various catalytic transformations [6]. Beller et al. reported the Mn(Ⅰ)-catalyzed hydrogenation of various polar functional groups in 2016 [7]. From then on, reduction reactions catalyzed by manganese complexes were studied but most of them are non-enantioselective reduction reactions [8-17]. Up to now, fewer chiral manganese catalysts are used for asymmetric reduction of ketones [18, 19]. In 2017, Zirakzadeh et al. demonstrated the first asymmetric transfer hydrogenations of ketones with manganese complexes of chiral PNP' pincer ligands, obtaining the chiral alcohols with conversions up to 96% and up to 86% ee [19a]. Recently, Mezzetti et al. reported the ATH of ketones catalyzed by bis(carbonyl) manganese(Ⅰ) complex, giving the corresponding chiral alcohols in high yields (up to > 99%) and with excellent enantioselectivities (90%–99% ee) [19g]. Compared with other non-precious metals such as Fe, Co, Ni, chiral Mn-based catalysts have been less developed. In our previous work, we developed the enantioselective catalytic reduction of various ketones by combining a series of chiral PxNy-type ligands (Fig. 1) with different transition metal complexes [20-25]. As part of our continuing study for novel asymmetric reduction catalysts, herein we described the chiral Mn(Ⅰ)-based catalyst containing of PxNy-type ligands for asymmetric transfer hydrogenation of ketones.

|
Download:
|
| Fig. 1. Chiral PxNy-type ligands. | |
Using iPrOH as hydrogen source and solvent, we investigated the asymmetric transfer hydrogenation catalyzed by [MnBr(CO)5] and various chiral PxNy-type ligands. Initially, we chose the acetophenone as the model reaction substrate. It could be seen that in the absence of chiral ligand, the reaction can hardly take place (Table 1, entry 1). Under the employed reaction conditions the catalyst generated in situ from [MnBr(CO)5] and chiral diiminodiphosphine ligand (R, R)-P2N2 (L1) afforded high conversion but low enantioselectivity (99% conversion and 21% ee, entry 2). With the catalyst generated in situ from [MnBr(CO)5] and chiral diaminodiphosphine ligand (R, R)-P2(NH)2 (L2), the acetophenone was asymmetric hydrogenated with 42% conversion and 71% ee (entry 3). When multidentate P3N4-type ligands L3 and L4 were used as chiral auxiliary, catalysts displayed good activity and moderate enantioselectivity (87% conversion and 65% ee, 66% conversion and 67% ee, repectively, entries 4 and 5). To our delight, the Mn(Ⅰ)-based catalyst containing chiral macrocyclic ligand (R, R, R', R')-CyP2N4 (L5), exhibited high activity and enantioselectivity for the ATH of acetophenone. In the presence of KOH, the ATH of acetophenone proceeded smoothly, affording the chiral product chiral product (S)-1-phenylethanol with 99% conversion and 81% ee in 2h at 75 ℃ with a substrate/catalyst (S/C) ratio of 50 (entry 6). However, as for the analogue of L5, chiral macrocyclic ligand (R, R, R', R')-CyP2(NH)4 (L6) gave the same high activity but moderate enantioselectivity under the same reaction conditions (99% conversion and 69% ee, entry 7). It is interesting that among the tested catalyst systems, the promoting effect of NH functions in the chiral PxNy-type ligand, which was notable feature in our previous results, does not work in this case. It could be seen that the reaction cannot proceed without metal precursor (entry 8). Furthermore, two other metal precursors MnCl2 and [Mn2(CO)10] were examined with the combination of L5. It showed that under the employed reaction conditions the catalyst generated in situ from MnCl2 and chiral macrocyclic ligand L5 was inactive for enantioselective reduction of acetophenone (entry 9). While the reaction catalyzed by [Mn2(CO)10]/L5 only gave the chiral product (S)-1-phenylethanol with 23% conversion and 47% ee (entry 10).
|
|
Table 1 Manganese-catalyzed ATH of acetophenone.a |
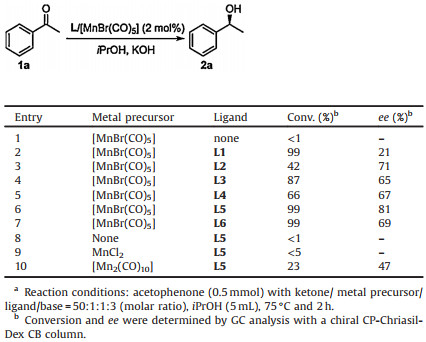
{kind=link}
Next, the catalytic performance of L5/[MnBr(CO)5] for the ATH of acetophenone were investigated. We examined the influence of the amount of bases and different temperatures (Please see the Tables in Supporting information). The enantioselective reduction was carried out with a catalyst loading of 2 mol%. It showed that at 75 ℃ after 2h of reaction time, no reaction took place in the absence of base. With increase of the amount of KOH, the activity of the catalyst also increased. When 2 equiv. of KOH were added, the acetophenone was enantioselectively hydrogenated with 63% conversion and 80% ee. Nearly complete conversion and 81% ee were achieved with a molar ratio of S: C: B (substrate: catalyst: KOH)=50:1:3. A high catalytic activity was observed upon increasing the temperature. At lower temperature (55 ℃), the catalytic activity dropped significantly and lower conversion (50%) was obtained. At 75 ℃ the yield of (S)-1-phenylethanol increased to 99% with 81% ee.
With the optimized reaction conditions in hand, the ATH of a wide range of aromatic ketones catalyzed by L5/[MnBr(CO)5] was examined. As shown in Scheme 1, various aromatic ketones were smoothly reduced under the conditions employed, affording the corresponding chiral aromatic alcohols with high yields and enantioselectivities. For aryl methyl ketones, it seems that there is no unambiguous correlation between the electron property of substituents on the phenyl ring and the yield of products. However, the enantioselectivities vary with the substitution position. Generally, the highest ee value is associated with ortho-substituted ketones. For example, 90% ee was obtained for the enantioselective hydrogenation of 2′-(trifluoromethyl)-acetophenone (o-1d), which is 12%, 15% higher than its meta-and para-substituted analogues (m-1d, p-1d), respectively. These results are similar to our previous results of iron-catalyzed asymmetric reduction of ketones [24]. Notably, the Mn(Ⅰ)-based catalyst is robust for the hydrogenation of bulky steric ketones, such as naphthyl ethanones (1m, 1n) and 1, 1′-diphenylpropan-2-one (1o). For instance, the ATH of 1-naphthyl ethanone (1n) proceeded smoothly at 75 ℃, giving the corresponding chiral alcohols with 98% yield and 95% ee in 2h. Furthermore, high enantioselectivities (85%–93% ee) were obtained with α-substituted acetophenones, including those bearing ethyl, propyl, butyl, and sterically more demanding tertbutyl groups (1p-1r, 1t). As for the ATH of α-isopropyl-phenylmethanone (1s), more KOH and longer reaction time were necessary to give high yield (S: C: B=50:1:5, 6h, 94% yield, 79% ee). The enantioselective reduction of diaryl ketones, such as 2-methylbenzophenone (1v) and (2-chlorophenyl)phenyl-methanone (1w), proceeded smoothly, affording the corresponding chiral products with 96% yield and moderate enantioselectivity. However, the L5/[MnBr(CO)5] catalyst failed to catalyze the ATH of dialkyl ketones under the employed conditions. It showed that the Mn(Ⅰ)-based catalyst is more suitable for ketones containing aromatic ring structure.

|
Download:
|
| Scheme 1. Manganese-catalyzed ATH of aromatic ketones. Reaction conditions: Ketones 0.5 mmol, ketone/[MnBr(CO)5]/L5/KOH=50:1:1:3 ( molar ratio), iPrOH (5 mL), 75 ℃. Yields were determined by 1H NMR analysis of the crude reaction mixture using 1, 3, 5-trimethoxybenzene as an internal standard. ee were determined by GC or HPLC analysis. aATH at 65 ℃. bKetone/[MnBr(CO)5]/L5/KOH=50:1:1:2 ( molar ratio). cKetone/[MnBr(CO)5]/L5/KOH=50:1:1:5 ( molar ratio). d(R)-Product, ligand (S, S, S', S')-L5 was used. | |
{kind=link}
Apart from aromatic ketones, the Mn(Ⅰ)-based catalyst L5/[MnBr(CO)5] was also examined to catalyze the ATH of heteroaromatic ketones to the corresponding chiral alcohols with 2 mol% of catalyst loading in the presence of 6 mol% KOH in iPrOH at 75 ℃ (Scheme 2). It could be seen that the tested heteroaromatic ketones were readily hydrogenated to yield the corresponding alcohols with high conversions and medium to high enantioselectivities. Notably, 3-acetyl-2, 5-dimethylthiophene (3c) is enantioselectively reduced to chiral thiophene alcohol with 94% yield and 91% ee in 3h. While furan derivatives were used as substrates, the catalytic system is inactive. It indicated that under the reaction conditions the Mn(Ⅰ)-based catalyst L5/[MnBr(CO)5] could not effectively activate the furan derivatives.

|
Download:
|
| Scheme 2. Mn(Ⅰ)-catalyzed ATH of heteroaromatic ketones. Reaction conditions: Ketones 0.5 mmol, ketone/[MnBr(CO)5]/L5/KOH=50:1:1:3 ( molar ratio), iPrOH (5 mL), 75 ℃. Yields were determined by 1H NMR analysis of the crude reaction mixture using 1, 3, 5-trimethoxybenzene as an internal standard. ee were determined by GC analysis with a chiral CP-Chriasil-Dex CB column. a(R)-Product, (S, S, S', S')-L5 ligand was used. | |
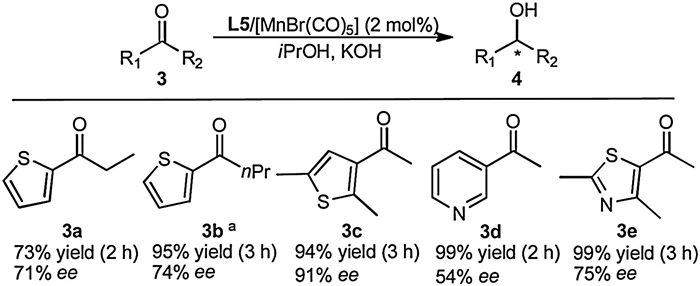{kind=link}
In summary, we have developed an efficient chiral Mn(Ⅰ)-based catalyst for asymmetric transfer hydrogenation of various aromatic ketones to obtain optically active alcohols. Using a combination of chiral macrocyclic ligand with [MnBr(CO)5] as the catalyst, prochiral ketones could be enantioselectively hydrogenated to corresponding chiral alcohol with up to 99% yield and up to 95% ee. Further investigations on the reaction mechanism are currently ongoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentWe are grateful to the National Natural Science Foundation of China (No. 21673190) for financial support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.023.
| [1] |
(a) J.G. de Vries, C.J. Elsevier, The Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2006; (b) T. Ikariya, Bifunctional molecular catalysis, in: T. Ikariya, M. Shibasaki (Eds.), Topics in Organometallic Chemistry, Springer-Verlag, Berlin, 2011 pp. 31; (c) Y.Y. Li, S.L. Yu, W.Y. Shen, et al., Acc. Chem. Res. 48 (2015) 2587-2598; (d) R.H. Morris, Acc. Chem. Res. 48 (2015) 1494-1502; (e) F. Foubelo, C. Najera, M. Yus, Tetrahedron Asymmetry 26 (2015) 769-790; (f) T. Ohkuma, N. Arai, Chem. Rec. 16 (2016) 2801-2819. |
| [2] |
A. Matsunami, Y. Kayaki, Tetrahedron Lett. 59 (2018) 504-513. DOI:10.1016/j.tetlet.2017.12.078 |
| [3] |
D. Wang, D. Astruc, Chem. Rev. 115 (2015) 6621-6686. |
| [4] |
M.J. Palmer, M. Wills, Tetrahedron Asymmetry 10 (1999) 2045-2061. DOI:10.1016/S0957-4166(99)00216-5 |
| [5] |
(a) F. Kallmeier, R. Kempe, Angew. Chem. Int. Ed. 57 (2018) 46-60; (b) D. Wei, C. Darcel, Chem. Rev. 119 (2019) 2550-2610; (c) I. Bauer, H.J. Knolker, Chem. Rev. 115(2015) 3170-3387; (d) H. Pellissier, Coord. Chem. Rev. 386 (2019) 1-31; (e) W.Y. Ai, R. Zhong, X.F. Liu, et al., Chem. Rev. 119 (2019) 2876-2953; (f) H. Pellissier, Coord. Chem. Rev. 360 (2018) 122-168; (g) H. Pellissier, H. Clavier, Chem. Rev. 114 (2014) 2775-2823; (h) S. Chakraborty, P. Bhattacharya, H.G. Dai, et al., Acc. Chem. Res. 48 (2015) 1995-2003. |
| [6] |
(a) N. Gorgas, K. Kirchner, Acc. Chem. Res. 51 (2018) 1558-1569; (b) Y.Y. Hu, B.W. Zhou, C.Y. Wang, Acc. Chem. Res. 51 (2018) 816-827; (c) W. Liu, J.T. Groves, Acc. Chem. Res. 48 (2015) 1727-1735; (d) A. Nodzewska, A. Wadolowska, M. Watkinson, Coord. Chem. Rev. 382 (2019) 181-216; (e) D.A. Valyaev, G. Lavigne, N. Lugan, Coord. Chem. Rev. 308 (2016) 191-235; (f) B. Qiu, C.G. Xia, W. Sun, Chin. Chem. Lett. 30 (2019) 698-701. |
| [7] |
S. Elangovan, C. Topf, S. Fischer, et al., J. Am. Chem. Soc. 138 (2016) 8809-8814. DOI:10.1021/jacs.6b03709 |
| [8] |
(a) S. Elangovan, M. Garbe, H. Jiao, et al., Angew. Chem. Int. Ed. 55 (2016) 15364-15368; (b) V. Papa, J.R. Cabrero-Antonino, E. Alberico, et al., Chem. Sci. 8 (2017) 3576-3585; (c) R. van Putten, E. Uslamin, M. Garbe, et al., Angew. Chem. Int. Ed. 56 (2017) 7531-7534; (d) M. Perez, S. Elangovan, A. Spannenberg, et al., ChemCatChem 10 (2017) 83-86. |
| [9] |
(a) F. Kallmeier, T. Irrgang, T. Dietel, et al., Angew. Chem. Int. Ed. 55 (2016) 11806-11809; (b) F. Freitag, T. Irrgang, R. Kempe, J. Am. Chem. Soc. 141 (2019) 11677-11685. |
| [10] |
A. Kaithal, M. Holscher, W. Leitner, Angew. Chem. Int. Ed. 57 (2018) 13449-13453. DOI:10.1002/anie.201808676 |
| [11] |
(a) A. Bruneau-Voisine, D. Wang, T. Roisnel, et al., Catal. Commun. 92 (2017) 1-4; (b) H.R. Li, D. Wei, A. Bruneau-Voisine, et al., Organometallics 37 (2018) 1271-1279; (c) D. Wei, A. Bruneau-Voisine, T. Chauvin, et al., Adv. Synth. Catal. 360 (2018) 676-681; (d) A. Bruneau-Voisine, D. Wang, V. Dorcet, et al., Org. Lett. 19 (2017) 3656-3659. |
| [12] |
(a) N.A. Espinosa-Jalapa, A. Nerush, L.J.W. Shimon, et al., Chem. Eur. J. 23 (2017) 5934-5938; (b) A. Kumar, T. Janes, N.A. Espinosa-Jalapa, et al., Angew. Chem. Int. Ed. 57 (2018) 12076-12080; (c) Y.Q. Zou, S. Chakraborty, A. Nerush, et al., ACS Catal. 8 (2018) 8014-8019; (d) U.K. Das, A. Kumar, Y. Ben-David, et al., J. Am. Chem. Soc. 141 (2019) 12962-12966. |
| [13] |
(a) M. Glatz, B. Stoger, D. Himmelbauer, et al., ACS Catal. 8 (2018) 4009-4016; (b) F. Bertini, M. Glatz, N. Gorgas, et al., Chem. Sci. 8 (2017) 5024-5029; (c) S. Weber, B. Stoger, K. Kirchner, Org. Lett. 20 (2018) 7212-7215. |
| [14] |
M.B. Widegren, M.L. Clarke, Org. Lett. 20 (2018) 2654-2658. DOI:10.1021/acs.orglett.8b00864 |
| [15] |
(a) K. Ganguli, S. Shee, D. Panja, et al., Dalton Trans. 48 (2019) 7358-7366; (b) N.V. Shvydkiy, O. Vyhiyskyi, Y.V. Nelyubina, et al., ChemCatChem 11 (2019) 1602-1605; (c) O. Martinez-Ferrate, C. Werle, G. Francio, et al., ChemCatChem 10 (2018) 4514-4518; (d) A. Dubey, S.M.W. Rahaman, R.R. Fayzullin, et al., ChemCatChem 11 (2019) 3844-3852; (e) R. van Putten, J. Benschop, V.J. de Munck, et al., ChemCatChem 11 (2019) 5232-5235. |
| [16] |
(a) Z.H. Shao, Y. Li, C.G. Liu, et al., Nat. Commun. 11 (2020) 591; (b) Y.J. Wang, L. Zhu, Z.H. Shao, et al., J. Am. Chem. Soc. 141 (2019) 17337-17349. |
| [17] |
Z.L. Wang, L. Chen, G.L. Mao, et al., Chin. Chem. Lett. 31 (2020) 1890-1894. DOI:10.1016/j.cclet.2020.02.025 |
| [18] |
(a) M.B. Widegren, G.J. Harkness, A.M.Z. Slawin, et al., Angew. Chem. Int. Ed. 56 (2017) 5825-5828; (b) M. Garbe, K. Junge, S. Walker, et al., Angew. Chem. Int. Ed. 56 (2017) 11237-11241; (c) L.L. Zhang, Y.T. Tang, Z.B. Han, et al., Angew. Chem. Int. Ed. 58 (2019) 4973-4977; (d) A. Passera, A. Mezzetti, Adv. Synth. Catal. 361 (2019) 4691-4706; (e) F. Ling, J.C. Chen, S.F. Nian, et al., Synlett 31 (2020) 285-289; (f) M. Garbe, Z.H. Wei, B. Tannert, et al., Adv. Synth. Catal. 361 (2019) 1913-1920; (g) M.B. Widegren, M.L. Clarke, Catal. Sci. Technol. 9 (2019) 6047-6058; (h) L.L. Zhang, Z. Wang, Z.B. Han, et al., Angew. Chem. Int. Ed. 59 (2020) 15565-15569; (i) X.C. Ma, Z.Q. Zuo, G.X. Liu, et al., ACS Omega 2 (2017) 4688-4692; (j) X.X. Yang, C.Y. Wang, Chem. Asian J. 13 (2018) 2307-2315. |
| [19] |
(a) A. Zirakzadeh, S.R.M.M. de Aguiar, B. Stoger, et al., ChemCatChem 9 (2017) 1744-1748; (b) F. Ling, H.C. Hou, J.C. Chen, et al., Org. Lett. 21 (2019) 3937-3941; (c) K.Z. Demmans, M.E. Olson, R.H. Morris, Organometallics 37 (2018) 4608-4618; (d) D. Wang, A. Bruneau-Voisine, J.B. Sortais, Catal. Commun. 105 (2018) 31-36; (e) J. Schneekonig, K. Junge, M. Beller, Synlett 30 (2019) 503-507; (f) R. van Putten, G.A. Filonenko, A.G. de Castro, et al., Organometallics 38 (2019) 3187-3196; (g) A. Passera, A. Mezzetti, Angew. Chem. Int. Ed. 59 (2020) 187-191. |
| [20] |
J.X. Gao, T. Ikariya, R. Noyori, Organometallics 15 (1996) 1087-1089. DOI:10.1021/om950833b |
| [21] |
J.X. Gao, H. Zhang, X.D. Yi, et al., Chirality 12 (2000) 383-388. DOI:10.1002/(SICI)1520-636X(2000)12:5/6<383::AID-CHIR15>3.0.CO;2-C |
| [22] |
M. Tao, F. Wu, T. Li, et al., Chin. Chem. Lett. 28 (2017) 97-100. DOI:10.1016/j.cclet.2016.05.028 |
| [23] |
S.L. Yu, W.Y. Shen, Y.Y. Li, et al., Adv. Synth. Catal. 354 (2012) 818-822. DOI:10.1002/adsc.201100733 |
| [24] |
Y.Y. Li, S.L. Yu, X.F. Wu, et al., J. Am. Chem. Soc. 136 (2014) 4031-4039. DOI:10.1021/ja5003636 |
| [25] |
D. Zhang, E.Z. Zhu, Z.W. Lin, et al., Asian J. Org. Chem. 5 (2016) 1323-1326. DOI:10.1002/ajoc.201600358 |