 2021, Vol. 32
2021, Vol. 32 


b Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
Sulfoxonium ylides have emerged as versatile building blocks in a plethora of annulation reactions by relying on their distinctive ambiphilic behaviors [1]. In this regard, many privileged carbo-and heterocyclic systems have been constructed hitherto by coupled with functionalized multiple C–X bonds (X = C, N, O, etc.) in an effective and selective fashion [2]. Meanwhile, enamides, behaving as the stable enamine counterparts, represent powerful structural motifs with wide applications in asymmetric reactions and natural product syntheses [3]. The electronic feature by decorating amide functionalities on olefins endows enamides with both nucleophilic and electrophilic characters to participate in various transformations [4]. Herein we report a silver-catalyzed [4+1] annulation [5] of secondary enamides with α-carbonyl sulfoxonium ylides, offering a practical route to access valuable multi-functionalized oxazoline derivatives.
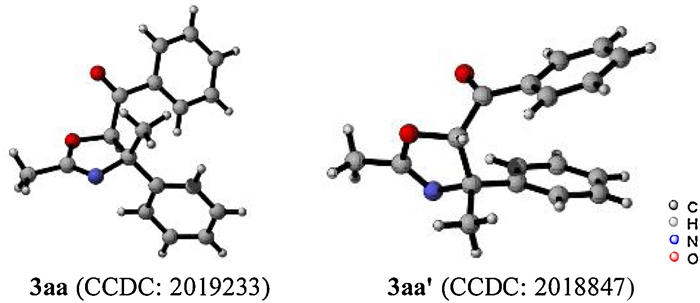
Oxazolines are important constituents embedded in a myriad of bioactive natural products and pharmaceuticals, such as aminorex, tambromycin, leptazolines A–D, tucatinib. [6]. In addition, these architectures frequently appear in chiral ligands such as renowned bis(oxazoline) and phosphinooxazoline families [7]. Therefore, the fascination with those molecules stimulates substantial interests of synthetic chemists over the past few decades (Scheme 1a). Classic approaches to build oxazoline skeletons include cyclodehydration of α-hydroxy amides (route a) [8], condensation of 1, 2-aminoalcohols with nitriles or carbonyls (route b) [9], and aldol condensation of isocyanoacetates with carbonyls (route c) [10]. Nevertheless, these pioneering contributions suffer from tedious procedures for preparing the starting materials and limited substitution patterns on the products. In response to these issues, a few elegant strategies have been reported in recent years. Tepe and co-workers accomplished the one-pot synthesis of tri-substituted oxazolines from sulfur ylides and acyl imines precursors (route d) [11]. Brachet and co-workers reported the visible light-induced annulation of benzoyl azides with alkenes to construct the oxazoline cores (route e) [12]. Intramolecular cyclization using allylic amides or β-amino ketones as the precursors also paved a way for the straightforward synthesis of di-and tri-substituted oxazolines (routes f and g) [13]. To our knowledge, construction of tetra-substituted oxazolines starting from readily accessible precursors remains a challenge and to be explored [14]. Given the significance of oxazolines and our interest in the enamide chemistry [15], we demonstrate the complement for the diversity-oriented synthesis of tetra-substituted oxazolines via the heteroannulation of enamides with α-carbonyl sulfoxonium ylides (Scheme 1b). Furthermore, the versatility of this protocol is illustrated by a series of useful transformations of the product.

|
Download:
|
| Scheme 1. Synthetic approaches to oxazolines. | |
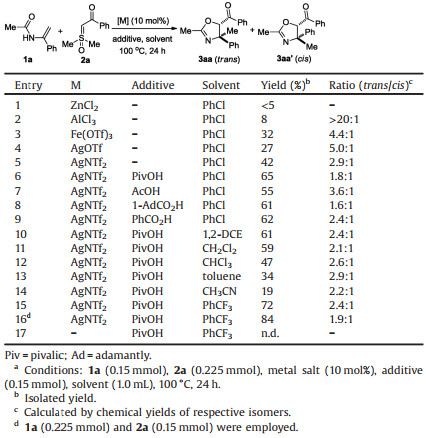
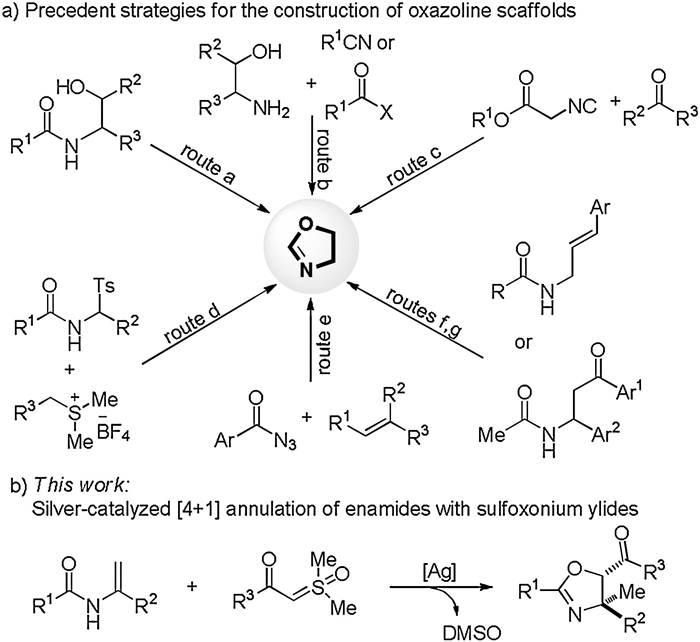
Initially, we selected N-(1-phenylvinyl)acetamide (1a) and sulfoxonium ylide (2a) as the model substrates to screen the reaction parameters (Table 1). The reactivity were first evaluated with a range of representative Lewis acids, among which AgNTf2 was the optimal catalyst (entries 1–5). Moderate diastereoselectivities were observed in all cases and, fortunately, two diastereomers could be readily separated by silica gel column chromatography. The relative stereochemistry of both isomers was unambiguously confirmed by X-ray crystallography (Fig. 1, for details, see Supporting information). The addition of PivOH remarkably increased the efficiency of the reaction (entry 6). Other acids led to diminished yields (entries 7–9). An extensive screening of solvents indicated that benzotrifluoride was the best medium (entries 10–15). It should be noted that the reaction was incompatible with other polar solvents, such as tetrahydrofuran, EtOAc, DMF and EtOH. Considering the partial decomposition of enamides in the catalytic system, we employed 2a as the limiting reagent and the products 3aa/ 3aa' were isolated in 84% combined yield (entry 16). A control experiment confirmed the necessity of the metal catalyst for the reaction to occur (entry 17).
|
|
Table 1 Optimization of the reaction conditions. |
{kind=link}

|
Download:
|
| Fig. 1. X-ray crystal structures of 3aa and 3aa'. | |
{kind=link}
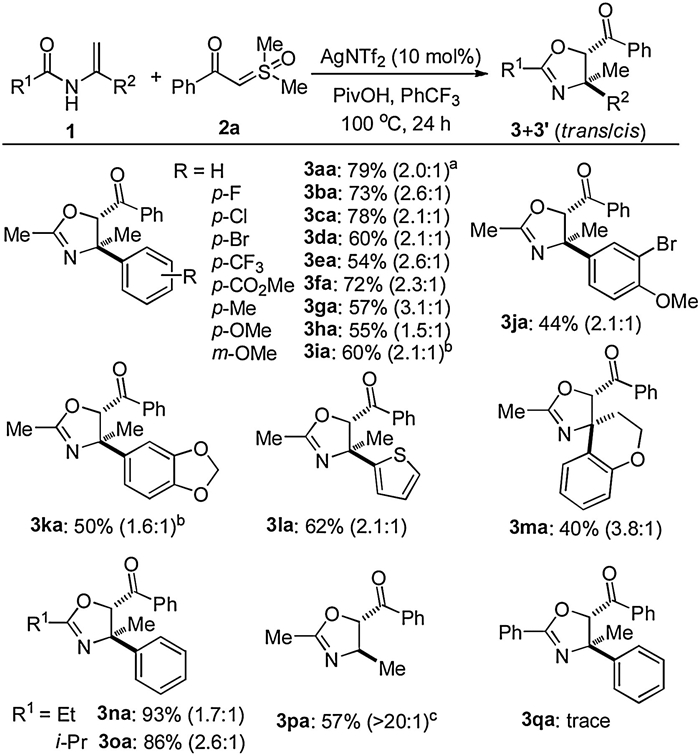
With optimized reaction conditions in hand, a multitude of enamides were evaluated for the tetra-substituted oxazolines syntheses (Scheme 2). The robustness of this method was demonstrated by a 1.0 mmol scale-up reaction, affording the annulation products in 79% yield (3aa). Introduction of various functional groups into the benzene ring was well tolerated, while keeping the halides, methyl, trifluoromethyl, ester, and acetal groups intact (3ba‒ ka). The electronic property had no apparent influence on the efficiency. Heteroaryl substrate bearing thienyl moiety successfully participated in the reaction to generate the targeted product in a promising yield (3la). Notably, biologically relevant enamide derived from chromanone led to the formation of spirocyclic oxazoline in an acceptable yield (3ma). In addition, the acetyl substituent on the nitrogen atom in enamides could be replaced with propionyl and isobutyryl moieties, and the corresponding products were provided in high yields (3na and 3oa). N-Vinylacetamide was subjected to the standard conditions, affording the tri-substituted oxazoline in a highly diasteroselective manner (3pa). The observation gave rise to the hint that diasteroselectivity in the transformation largely relied on substrate control with different steric factor. Unfortunately, only trace amount of product 3qa was detected by employing N-benzoyl substituted enamide as the substrate under the current conditions.

|
Download:
|
| Scheme 2. Substrate scope with respect to enamides. Reaction was performed by using 1 (0.225 mmol), 2a (0.15 mmol), AgNTf2 (0.015 mmol), and PivOH (0.15 mmol) in PhCF3 at 100 ℃ for 24 h. Combined yields of isolated products are given. Diastereomeric ratios in parentheses were calculated by yields of respective isomers. a 1.0 mmol scale.b At 110 ℃. c At 60 ℃, diastereomeric ratio in parentheses was determined by1H NMR analysis. | |
{kind=link}
Subsequently, we turned our attention to investigate the generality and limitation of sulfoxonium ylides using enamide 1c bearing a chloro substituent as a synthetic handle for potential modification. As shown in Scheme 3, ylides decorated with both electron-deficient and -rich substituents at ortho, meta-, and para-positions of the phenyl ring proved to be suitable partners for annulation, affording the desired products in 64%–97% combined yields (3cb‒ ci). Likewise, the process could be expanded to other arenes, including thienyl and naphthyl substituted sulfoxonium ylides, delivering the corresponding oxazolines (3cj and 3ck). However, alkyl substituted ylide with the example of benzyl group was found to be incompatible with the reaction at the current stage (3cl).

|
Download:
|
| Scheme 3. Substrate scope with respect to sulfoxonium ylides. The conditions are the same as those in Scheme 2. | |
{kind=link}
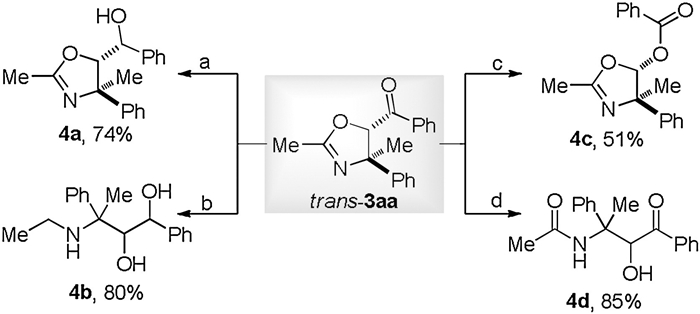
To illustrate the synthetic utility of our new protocol, we subsequently carried out some useful manipulations of the resultant tetra-substituted oxazoline 3aa via established strategies (Scheme 4). Reduction of tethered ketone moiety under NaBH4 gave the alcohol 4a in 74% yield. Treatment of 3aa with diisobutylaluminium hydride (DIBAL-H) resulted in the ring-cleavage reaction of oxazoline, leading to the valuable 3-amino-1, 2-diol 4b in 80% yield. The benzoyl functionality in 3aa underwent Baeyer-Villiger oxidation in the presence of m-chloroperbenzoic acid (mCPBA) and the corresponding ester 4c was obtained in a satisfactory yield. Moreover, compound 3aa could be easily hydrolyzed to versatile α-hydroxyl-β-amino ketone 4d while exposing to the concentrated HCl solution.

|
Download:
|
| Scheme 4. Product diversification. (a) NaBH4, MeOH, r.t., 3 h. (b) DIBAL-H, toluene, −78 ℃ to r.t., 2 h. (c) mCPBA, NaHCO3, DCM, r.t., 7 h. (d) HCl, THF, r.t., 2 h. | |
{kind=link}
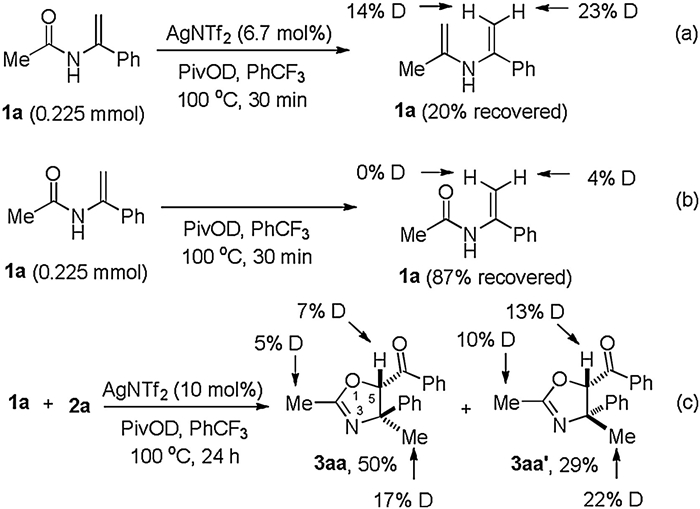
Finally, we conducted several preliminary D-labelling experiments to gain mechanistic insight into the annulation (Scheme 5). Performing the reaction in the absence of 2a with the addition of PivOD led to a significant deuterium incorporation in the recovered 1a (Scheme 5a). However, no deuterated 1a was observed when silver salt was absent (Scheme 5b). Moreover, under the standard conditions by using PivOD, approximately 17% and 22% deuterium were incorporated into the C4 positions of 3aa and 3aa', respectively (Scheme 5c). These findings implied that the reversible tautomerization between enamide and in situ-generated imine was facilitated by Lewis acid, yet which played a detrimental role for the stabilization of enamide.

|
Download:
|
| Scheme 5. Deuterium-labelling experiments. | |
{kind=link}
On the basis of preliminary results and literature precedents concerning Lewis acid-promoted transformations with enamides or ylides [16], a plausible mechanism for Ag-catalyzed [4+1] heteroannulation is depicted inScheme 6. The catalytic cycle starts from coordination of the metal center of silver catalyst with the oxygen atom of enamide 1a. The presence of PivOH enhances the electrophilic reactivity of the enamide through the delocalization of the lone-pair electrons of nitrogen atom into olefinic π-conjugation system. Nucleophilic addition of sulfoxonium ylide 2a into iminium carbon of species II produces species Ⅲ. Deprotonation induced by NTf2 anion and tautomerization gives species Ⅳ, which undergoes elimination of DMSO to form the final product. The diastereoselectivity in the annulation can be rationalized by the conformational analysis of species Ⅳ, in which conformer A is favorable due to its relatively less steric hindrance.

|
Download:
|
| Scheme 6. Proposed reaction mechanism. | |
{kind=link}
In conclusion, we have disclosed a practical route for the construction of diversely functionalized oxazoline derivatives readily assembled by Ag-catalyzed [4+1] heteroannulation of enamides with α-carbonyl sulfoxonium ylides. The protocol is compatible with various secondary enamides with good functional groups tolerance. Importantly, this approach brings a reliable avenue to the challenge for the streamline synthesis of tetra-substituted oxazolines. Moreover, some synthetic transformations into valuable building blocks highlight the potential of this procedure. We anticipate an enantioselective version of this reaction and further applications of enamides in heterocycles syntheses in the future.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful to the National Natural Science Foundation of China (No. 21702106), the Natural Science Foundation of Jiangsu Province (No. BK20170967), and the Start-up Grant from Nanjing Tech University (No. 39839101) for financial support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.007.
| [1] |
(a) X.L. Sun, Y. Tang, Acc. Chem. Res. 41 (2008) 937-948; (b) L.Q. Lu, J.R. Chen, W.J. Xiao, Acc. Chem. Res. 45 (2012) 1278-1293; (c) G. Li, L. Wang, Y. Huang, Chin. J. Org. Chem. 33 (2013) 1900-1918; (d) L.Q. Lu, T.R. Li, Q. Wang, W.J. Xiao, Chem. Soc. Rev. 46 (2017) 4135-4149; (e) D. Kaiser, I. Klose, R. Oost, J. Neuhaus, N. Maulide, Chem. Rev. 119 (2019) 8701-8780. |
| [2] |
(a) Q. Wang, T.R. Li, L.Q. Lu, et al., J. Am. Chem. Soc. 138 (2016) 8360-8363; (b) Y. Xu, G. Zheng, X. Yang, X. Li, Chem. Commun. 54 (2018) 670-673; (c) X. Wu, H. Xiong, S. Sun, J. Cheng, Org. Lett. 20 (2018) 1396-1399; (d) D. Clare, B.C. Dobson, P.A. Inglesby, C. Aïssa, Angew. Chem. Int. Ed. 58 (2019) 16198-16202; (e) Z. Tang, Y. Zhou, Q. Song, Org. Lett. 21 (2019) 5273-5276; (f) X. Chen, M. Wang, X. Zhang, X. Fan, Org. Lett. 21 (2019) 2541-2545; (g) S. Hu, S. Du, Z. Yang, L. Ni, Z. Chen, Adv. Synth. Catal. 361 (2019) 3124-3136; (h) Z. Shen, C. Pi, X. Cui, Y. Wu, Chin. Chem. Lett. 30 (2019) 1374-1378; (i) T.B. Hua, C. Xiao, Q.Q. Yang, J.R. Chen, Chin. Chem. Lett. 31 (2020) 311-323; (j) Y. Kommagalla, S. Ando, N. Chatani, Org. Lett. 22 (2020) 1375-1379; (k) S. Zhu, K. Shi, H. Zhu, et al., Org. Lett. 22 (2020) 1504-1509. |
| [3] |
(a) R. Matsubara, S. Kobayashi, Acc. Chem. Res. 41 (2008) 292-301; (b) D.R. Carbery, Org. Biomol. Chem. 6 (2008) 3455-3460; (c) K. Gopalaiah, H.B. Kagan, Chem. Rev. 111 (2011) 4599-4657. |
| [4] |
(a) M.X. Wang, Chem. Commun. 51 (2015) 6039-6049; (b) X.M. Xu, L. Zhao, J. Zhu, M.X. Wang, Angew. Chem. Int. Ed. 55 (2016) 3799-3803; (c) J. Wu, C. Zhao, J. Wang, J. Am. Chem. Soc. 138 (2016) 4706-4709; (d) M.N. Zhao, L. Yu, R.R. Hui, et al., ACS Catal. 6 (2016) 3473-3477. |
| [5] |
(a) J.R. Chen, X.Q. Hu, L.Q. Lu, W.J. Xiao, Chem. Rev. 115 (2015) 5301-5365; (b) C. Zhu, Y. Ding, L.W. Ye, Org. Biomol. Chem. 13 (2015) 2530-2536; (c) P. Sivaguru, S. Cao, K.R. Babu, X. Bi, Acc. Chem. Res. 53 (2020) 662-675. |
| [6] |
(a) A.W. Goering, R.A. McClure, J.R. Doroghazi, et al., ACS Cent. Sci. 2 (2016) 99-108; (b) J.B. Neupana, R.P. Neupane, Y. Luo, et al., Org. Lett. 21 (2019) 8449-8453; (c) J. Maier, F.P. Mayer, S.D. Brandt, H.H. Sitte, ACS Chem. Neurosci. 9 (2018) 2484-2502; (d) L. Liu, L. Yin, H. Bian, N. Zhang, Chin. Chem. Lett. 31 (2020) 501-504. |
| [7] |
(a) G. Helmchen, A. Pfaltz, Acc. Chem. Res. 33 (2000) 336-345; (b) G.C. Hargaden, P.J. Guiry, Chem. Rev. 109 (2009) 2505-2550. |
| [8] |
(a) P. Wipf, C.P. Miller, Tetrahedron Lett. 33 (1992) 907-910; (b) A.J. Phillips, Y. Uto, P. Wipf, M.J. Reno, D.R. Williams, Org. Lett. 2 (2000) 1165-1168. |
| [9] |
(a) S. Rajaram, M.S. Sigman, Org. Lett. 4(2002) 3399-3401; (b) T. Ohshima, T. Iwasaki, K. Mashima, Chem. Commun. (2006) 2711-2713; (c) M. Trose, F. Lazreg, M. Lesieur, C.S.J. Cazin, J. Org. Chem. 80 (2015) 9910-9914. |
| [10] |
(a) T. Saegusa, Y. Ito, H. Kinoshita, S. Tomita, J. Org. Chem. 36 (1971) 3316-3323; (b) F. Sladojevich, A. Trabocchi, A. Guarna, D.J. Dixon, J. Am. Chem. Soc. 133 (2011) 1710-1713. |
| [11] |
M.S.A. Mehedi, J.J. Tepe, J. Org. Chem. 84 (2019) 7219-7226. DOI:10.1021/acs.joc.9b00883 |
| [12] |
P. Bellotti, J. Brocus, F.E. Orf, et al., J. Org. Chem. 84 (2019) 6278-6285. |
| [13] |
(a) P.D. Morse, D.A. Nicewicz, Chem. Sci. 6 (2015) 270-274; (b) W.C. Gao, F. Hu, Y.M. Huo, et al., Org. Lett. 17 (2015) 3914-3917; (c) H. Wang, J. Zhang, J. Tan, et al., Org. Lett. 20 (2018) 2505-2508; (d) S.S. Chavan, B.D. Rupanawar, R.B. Kamble, A.M. Shelke, G. Suryavanshi, Org. Chem. Front. 5 (2018) 544-548; (e) H. Luo, Z. Yang, W. Lin, Y. Zheng, S. Ma, Chem. Sci. 9 (2018) 1964-1969. |
| [14] |
Y. He, C. Pi, Y. Wu, X. Cui, Chin. Chem. Lett. 31 (2020) 396-400. |
| [15] |
(a) S. Pankajakshan, Y.H. Xu, J.K. Cheng, M.T. Low, T.P. Loh, Angew. Chem. Int. Ed. 51 (2012) 5701-5705; (b) Z.Y. Shen, J.K. Cheng, C. Wang, et al., ACS Catal. 9 (2019) 8128-8135; (c) P. Shi, S. Li, L.M. Hu, et al., Chem. Commun. 55 (2019) 11115-11118; (d) R.H. Liu, Z.Y. Shen, C. Wang, T.P. Loh, X.H. Hu, Org. Lett. 22 (2020) 944-948; (e) S. Li, Q.C. Shan, L.M. Hu, X.Q. Ma, X.H. Hu, Chem. Commun. 56 (2020) 7969-7972. |
| [16] |
(a) B. Maji, S. Lakhdar, H. Mayr, Chem. Eur. J. 18 (2012) 5732-5740; (b) L. Yang, Q. Wen, F. Xiao, G.J. Deng, Org. Biomol. Chem. 12 (2014) 9519-9523; (c) X.M. Xu, C.H. Lei, S. Tong, J. Zhu, M.X. Wang, Org. Chem. Front. 5 (2018) 3138-3142; (d) Y. He, J. Lou, Z. Yu, Y.G. Zhou, Chem. Eur. J. 26 (2020) 4941-4946. |