 2021, Vol. 32
2021, Vol. 32 


b Department of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, United States;
c The State Key Laboratory of Chemical Oncogenomics, Guangdong Provincial Key Laboratory of Nano-Micro Materials Research, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen 518055, China
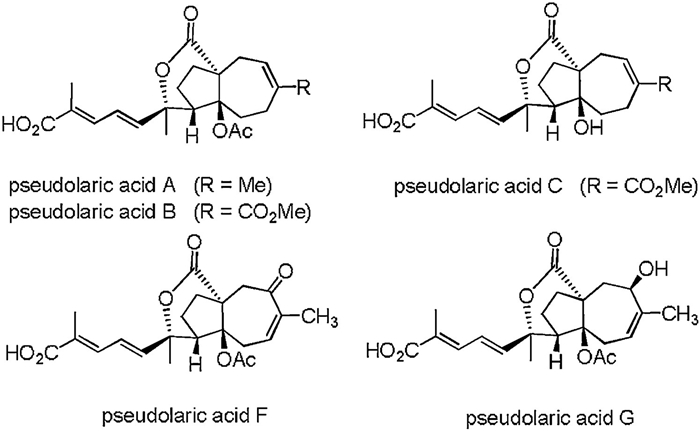
Pseudolaric acids are a class of novel deterpenoids isolated from the root bark of Pseudolarix kaempferi Gordon (pinaceae) by Chinese scientists in 1980's [1, 2]. To date, more than 20 pseudolaric acid analogues have been isolated successively [3-6], exhibiting significant cytotoxic activities against numerous tumor cell lines and strong antifungal activities [7-8]. Of them, pseudolaric acid B displays much higher activities than other pseudolaric acid members. As shown in Fig. 1, this family of compounds features a rare trans-fused [5–7]-bicyclic core, four contiguous stereocenters, and a rigid bridged ring structure substituted with an acetoxy group and a lactone at the ring junction.

|
Download:
|
| Fig. 1. Structures of pseudolaric acid A, B, C, F and G. | |
{kind=link}
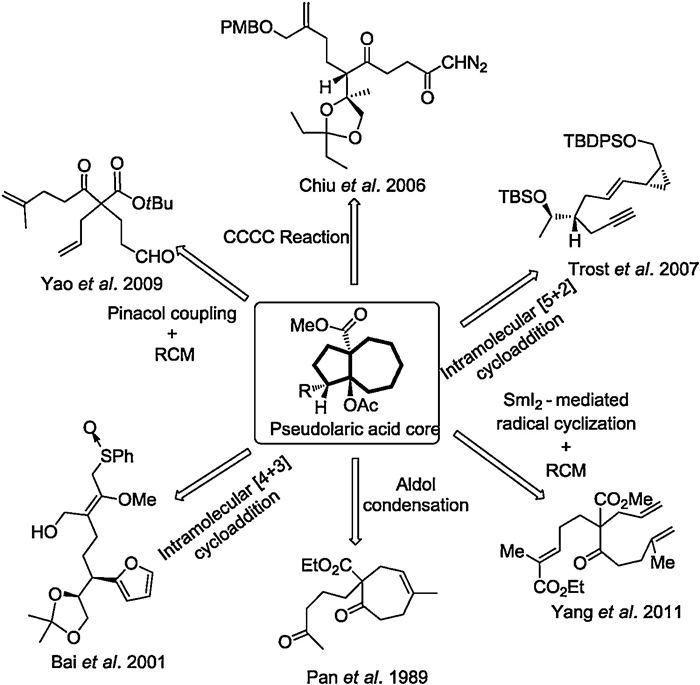
Due to their remarkable biological activities and intriguing structural skeleton, the pseudolaric acids have attracted considerable attention from the synthetic community. In 2006, Chiu group reported the first total synthesis of pseudolaric acid A, in which an Evans catalytic asymmetric aldol reaction and a rhodium-catalyzed intramolecular carbene cyclization cycloaddition cascade (CCCC) reaction were employed to construct the polycyclic framework (Fig. 2) [9]. Subsequently, Trost and coworkers disclosed the first total synthesis of pseudolaric acid B, enabled by a rhodium-catalyzed intramolecular [5+2] cyclization reaction to forge the bicyclic[5.3.0]decane skeleton and an intramolecular acyl radical cyclization to construct the lactone structure [10, 11]. Yang group completed a 16-steps synthesis of pseudolaric acid A in 2011, which exploited a samarium diiodide (SmI2)-mediated intramolecular radical cyclization and a ring-closing metathesis (RCM) reaction to construct the unique trans-substituted fused [5–7] ring system [12]. Besides, a number of synthetic studies were also developed to accomplish the synthesis of pseudolaric acids. As shown inFig. 2, Pan group developed a strategy using aldol condensation to produce the [5–7]-bicyclic skeleton of pseudolaric acids [13]. However, this method only forms cis-fused product. In 2001, Bai and coworkers took advantage of intramolecular Pummerer rearrangement and [4+3] cycloaddition reaction to construct the bicyclic[5.3.0]decane skeleton [14, 15]. Yao group also disclosed a strategy to assemble the [5–7]-bicyclic core structure, which involved pinacol coupling and RCM reaction [16]. Herein, we reported the enantioselective synthetic studies towards the key trans-fused [5–7]-bicyclic core skeleton of pseudolaric acid B, which featured the Sharpless asymmetric epoxidation, cyanide-opening reaction of the epoxide, and intramolecular [5+2] cycloaddition reaction as the key transformations.

|
Download:
|
| Fig. 2. The previous synthetic strategy of pseudolaric acids. | |
{kind=link}
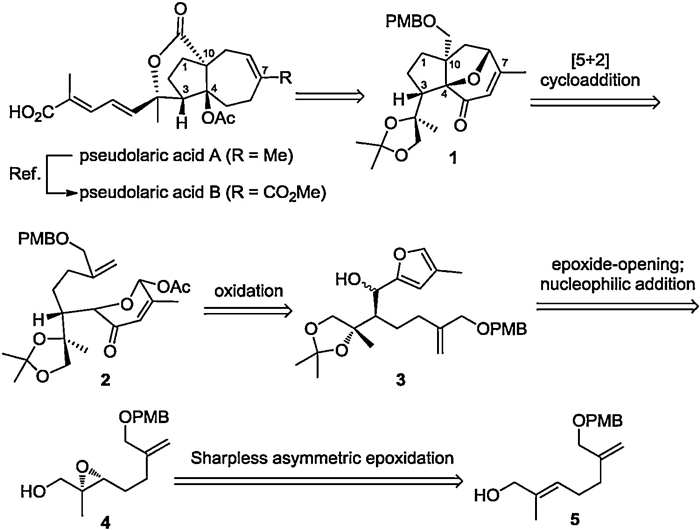
As depicted in Scheme 1, we envisioned that pseudolaric acid A and B could be synthesized from the key tricyclic skeleton 1 through selective reductive cleavage of bridged ether, oxidized lactonization, and late-stage Horner–Wadsworth–Emmons (HWE) reaction. As for tricyclic intermediate 1, it could be obtained via an intramolecular [5+2] cycloaddition reaction of pyrylium precursors2. This reaction would be the key step of our synthetic strategy to construct the trans-fused [5–7]-bicyclic core in pseudolaric acids. Pyrylium precursors2 could be assembled by oxidation of furan intermediates 3, which was envisioned to be constructed from epoxide 4 through a selective cyanide-opening reaction of epoxide and subsequent nucleophilic addition. Finally, epoxide 4 could be obtained from allyl alcohol 5 by Sharpless asymmetric epoxidation to introduce the initial stereocenter.

|
Download:
|
| Scheme 1. Retrosynthetic analysis. | |
{kind=link}
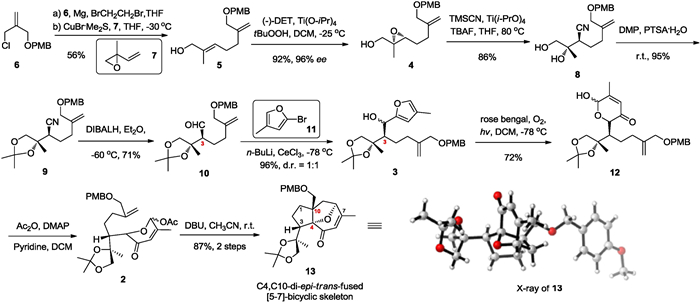
Our synthesis commenced from the known compound 3-chloro-2-benzyloxymethylpropene 6 which was steadily prepared from methallyl dichloride according to the reported procedure (Scheme 2) [9]. SN2' substitution reaction of Grignard reagent (in situ preparation from allyl chloride 6) and 2-methyl-2-vinyloxirane 7 under the catalysis of CuBr·MeS2 delivered the allyl alcohol 5 in 56% yield. To our delight, the subsequent Sharpless asymmetric epoxidation of allyl alcohol 5 proceeded smoothly to deliver the epoxide 4 in high yield and enantioselectivity (92%, 96% ee) [17-19]. Inspired by Konno group's report [20], we then attempted the cyanide-opening reaction of epoxide4. Treatment of 4 with trimethylsilyl cyanide (TMSCN) and anhydrous tetrabutylammonium fluoride (TBAF) in tetrahydrofuran could afford the β-hydroxy cyanide 8 in 51% yield, along with a large amount of starting material 4 remaining. We speculate that the low reactivity of epoxide may be responsible for the low yield and conversion. After screening a series of Lewis acid, we found that when treated with 2.0 equiv. of titanium tetraisopropanolate (Ti(O-iPr)4), the yield of β-hydroxy cyanide could be improved to 86%, which indicates this reaction could be significantly accelerated by Ti(O-iPr)4 (see the Supporting information for details). Protection of the dihydroxy moiety of compound 8 with 2, 2-dimethoxypropane (DMP) smoothly gave product 9 in 95% yield. Subsequent reduction of the cyano group with diisobutylaluminum hydride (DIBALH) produced aldehyde 10 in 71% yield. In the next nucleophilic addition step, the 4-methylfuryl lithium reagent was converted into a corresponding cerium reagent to weaken its basicity [21]. Therefore, the isomerization of C3 position in3 could be minimized. Compounds 3 were obtained as a mixture of two diastereoisomers (1:1 ratio) in 96% combined yield. According to the Magnus' procedure [22], unstable pyranenones12 were obtained in 72% yield, which went through acetyl protection immediately to give pyrylium precursors 2 in high yield. Without further purification, compounds 2 could be used directly in the next step.

|
Download:
|
| Scheme 2. Enantioselective synthesis of C4, C10-di-epi-trans-fused [5–7]-bicyclic skeleton. | |
{kind=link}
With the pyrylium precursors 2 in hand, we turned our attention to the key intramolecular [5+2] cycloaddition strategy to construct the trans-fused [5–7]-bicyclic skeleton (Scheme 2). However, under the activation of 1, 8-diazabicyclo[5, 4, 0]undec-7-ene (DBU) in acetonitrile [23-25], intramolecular [5+2] cycloaddition reaction took place only to form tricyclic product13 as a single isomer in 87% overall yields (2 steps), which contains opposite stereocenters at the ring junction position C4 and C10 compared with pseudolaric acids. Other reaction conditions, for example, treatment of a dilute solution of 12 in dichloromethane with trifluoroacetic acid [22], did not give corresponding cycloaddition product. The stereochemistry of13 was determined by the X-ray crystallographic analysis.
As the transition states (TS) shown in Scheme 3, we anticipate that the dihydroxy side chain in compound 2 as the larger group relative to the hydrogen atom should be disposed of in the equatorial position. Besides, the more favorable endo transition state of the pyrylium ylide and double bond would produce trans-fused [5–7]-bicyclic skeleton. According to the experiment results, TS-2 is presumably more favorable than TS-1, which affords 13 as the major product. It might be because that TS-2 has a less steric repulsion between the substituents compared with TS-1.

|
Download:
|
| Scheme 3. Transition state analysis. | |
{kind=link}
In summary, the C4, C10-di-epi-trans-fused [5–7]-bicyclic core of pseudolaric acid B, has been accomplished in 10 steps from the commercially available material methallyl dichloride. The key transformations of our strategy include the Sharpless asymmetric epoxidation, Ti(O-iPr)4-promoted cyanide-opening reaction of the epoxide, and intramolecular [5+2] cycloaddition. Further attempts of adjusting cycloaddition reaction to construct correct stereocenters and investigations to complete the total synthesis of pseudolaric acid B are underway.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos. 21302078, 21572089, 21732001, 21672017), the Program for Changjiang Scholars and the Innovative Research Team in Universities (PCSIRT: No. IRT_15R28), the State Key Basic Research Program of the PRC (No. 2018YFC0310900), Shenzhen Science and Technology Innovation Committee (No. JCYJ20180504165454447), Shenzhen Basic Research Program (No. 20180202) and the National Ten Thousand Talent Program (the Leading Talent Tier).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.023.
| [1] |
Z.L. Li, D.J. Pan, C.Q. Hu, Q.L. Wu, S.S. Yang, G.Y. Xu, Acta Chim. Sin. 40 (1982) 447-457. |
| [2] |
J.X. Yao, X.Y. Lin, Acta Chim. Sin. 40 (1982) 385-393. |
| [3] |
K. Hostettmann, A. Marston, M. Maillard, M. Hamburger, Nat. Prod. Lett. 9 (1996) 79. DOI:10.1080/10575639608043582 |
| [4] |
E. Li, A.M. Clark, C.D. Hufford, J. Nat. Prod. 58 (1995) 57-67. DOI:10.1021/np50115a007 |
| [5] |
X.C. Li, H.N. Eisonhly, A.C. Nimord, A.M. Clark, J. Nat. Prod. 62 (1998) 767-769. |
| [6] |
S.P. Yang, Y. Wu, J. Yue, M.J. Nat. Prod. 65 (2002) 1041-1044. DOI:10.1021/np0200010 |
| [7] |
V.K. Wong, P. Chiu, S.S. Chung, et al., Clin. Cancer Res. 11 (2005) 6002-6011. DOI:10.1158/1078-0432.CCR-05-0209 |
| [8] |
X.F. Gong, M.W. Wang, Z. Wu, S.I. Tashiro, S. Onodera, T. Ikejima, Exp. Mol. Med. 36 (2004) 551-556. DOI:10.1038/emm.2004.70 |
| [9] |
Z. Geng, B. Chen, P. Chiu, Angew. Chem. Int. Ed. 45 (2006) 6197-6200. DOI:10.1002/anie.200602056 |
| [10] |
B.M. Trost, J. Waser, A. Meyer, J. Am. Chem. Soc. 129 (2007) 14556-14557. DOI:10.1021/ja076165q |
| [11] |
B.M. Trost, J. Waser, A. Meyer, J. Am. Chem. Soc. 130 (2008) 16424-16434. DOI:10.1021/ja806724x |
| [12] |
T. Xu, C.C. Li, Z. Yang, Org. Lett. 13 (2011) 2630-2633. DOI:10.1021/ol200741j |
| [13] |
B.C. Pan, H.Y. Chang, G.L. Cai, Y.S. Guo, Pure Appl. Chem. 61 (1989) 389-392. DOI:10.1351/pac198961030389 |
| [14] |
Y.H. Hu, L.G. Ou, X.L. Wang, D.L. Bai, Chin. Chem. Lett. 10 (1999) 281-284. |
| [15] |
X.T. Jiang, L.G. Ou, D.M. Han, D.L. Bai, Chin. Chem. Lett. 12 (2001) 113-116. |
| [16] |
K. Cui, H. Liao, Z.J. Yao, Synthetic studies toward pseudolaric acid B, 11th Tetrahedron Symposium. Beijing: Tetrahedron Symposium, 2010: PSC113.
|
| [17] |
R.M. Hanson, K.B. Sharpless, J. Org. Chem. 51 (1986) 1922-1925. DOI:10.1021/jo00360a058 |
| [18] |
W. Wang, X. Zhang, Y. Zhao, et al., Chin. Chem. Lett. 29 (2018) 1872-1874. DOI:10.1016/j.cclet.2018.02.006 |
| [19] |
L. Liu, B. Wang, C. Bi, G. He, G. Chen, Chin. Chem. Lett. 29 (2018) 1113-1115. DOI:10.1016/j.cclet.2018.05.012 |
| [20] |
H. Konno, E. Toshiro, N. Hinoda, Synthesis 14 (2003) 2161-2164. |
| [21] |
T. Imamoto, Y. Sugiura, N. Takiyama, Tetrahedron Lett. 25 (1984) 4233-4236. DOI:10.1016/S0040-4039(01)81404-0 |
| [22] |
P. Magnus, L. Shen, Tetrahedron 55 (1999) 3553-3560. DOI:10.1016/S0040-4020(98)01162-4 |
| [23] |
M. Zhang, N. Liu, W.P. Tang, J. Am. Chem. Soc. 135 (2013) 12434-12438. DOI:10.1021/ja406255j |
| [24] |
G. Mei, X. Liu, C. Qiao, W. Chen, C.C. Li, Angew. Chem. Int. Ed. 54 (2015) 1754-1758. DOI:10.1002/anie.201410806 |
| [25] |
X. Liu, Y.J. Hu, J.H. Fan, et al., Org. Chem. Front. 5 (2018) 1217-1228. DOI:10.1039/C7QO01123G |