 2021, Vol. 32
2021, Vol. 32 


Coordination cages have attracted considerable attention due to their potential applications in metallasupramolecular materials and biomedical therapy [1-7]. Cages with distinct conformations such as globular, tetrahedral, and columnar shapes have been widely developed and lead to the diversity of features [8-13]. Among these architectures, the mechanically interlocked cages, in which two monomeric cages are interpenetrated with each other, offer a powerful tool to increase complexity of structures and functions [14-25]. The process of interlocking is entropically disfavored, while the gain of enthalpy via supramolecular interactions, such as hydrogen bonding, π-π interaction, and electrostatic interaction within the cages is prerequisite. In order to facilitate these interactions, the flexibility of ligand is normally required, whereby the adaptable feature can facilitate the aggregate between the two monomeric cages [26].
The cages made of aromatic amide ligands, reported by us and other groups, possess the flexible character due to the twisting of the amide moieties [27-31]. Following this, we hypothesised that an aromatic amide ligand long enough may also be prone to assemble into an interlocked cage via coordination. The target ligand L was therefore designed and synthesized by coupling of reported 4, 4′-diaminodiphenylmethylamine with commercially available nicotinic acid (Scheme S1 in Supporting information).
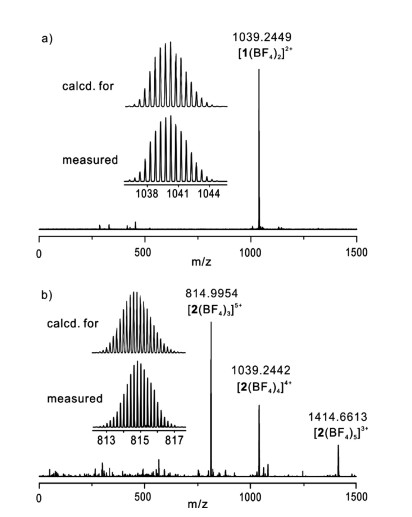
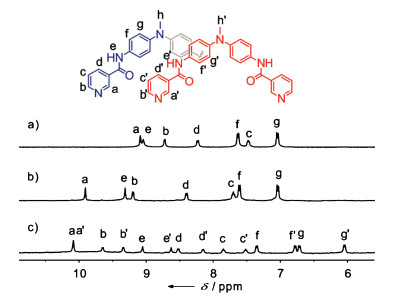
The monomeric cage 1 was initially formed by the reaction of L with [Pd(CH3CN)4](BF4)2 at 2:1 ratio in CD3CN for 2 h at 303 K (Scheme 1). ESI mass spectrum verified its structure of a M2L4 assembly (Fig. 1a). The observed peaks at m/z 1039.2 were assigned to the [1(BF4)2]2+ species. The 1H NMR spectrum of the solution also supported the formation of the metal complex 1: the pyridyl protons, in particular Ha and Hb, showed large downfield shifts, a characteristic of metal-ligand coordination (Figs. 2a and b), while the other aromatic protons displayed much smaller shifts. In addition, only one set of signals for each of the ligand's proton environments was presented for the cage 1, in agreement with its high symmetry of structure.

|
Download:
|
| Scheme 1. Synthesis of the monomeric and the dimeric cages. The reaction of L (2 equiv.) with [Pd(CH3CN)4](BF4)2 (1 equiv.) in acetonitrile at 303 K led to the formation of 1. Prolongation of the reaction at 333 K gave rise to the interlocked complex 2. | |
{kind=link}

|
Download:
|
| Fig. 1. The high-resolution ESI-MS of (a) the monomeric cage 1 and (b) the dimeric cage 2. | |
{kind=link}
Prolongation of the reaction at 333 K led to the quantitative formation of a new species accompanied by a splitting of all the NMR signals into two sets of equal intensity (Scheme 1 and Fig. 2c).

|
Download:
|
| Fig. 2. 1H NMR spectra of (a) the ligand L, (b) the monomeric cage 1 and (c) the dimeric cage 2 in CD3CN. | |
{kind=link}
The ESI mass analysis displayed a family of prominent signals at m/z 815.0, 1039.2 and 1414.7, which were assigned to the dimeric species [2(BF4)3]5+, [2(BF4)4]4+ and [2(BF4)5]3+, respectively (Fig. 1b). These results suggested that complex 1 converted into a more thermodynamically stable complex 2, as showed in Scheme 1.
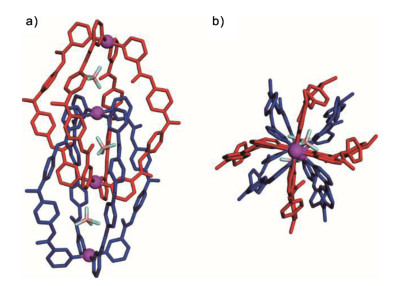
Single-crystal X-ray analysis unambiguously reveals the formation of the dimeric, interlocked cage 2 (Fig. 3). The interlocking produces three internal cavities of the structure with nearly equal volumes, whereby one tetrafluoroborate ion is encapsulated per cavity to balance the positive charges. Interestingly, in the solid state, the structure of 2 displays severe distortion compared to other reported interlocked cages [20-26]. Firstly, the tertiary aniline segments in the central of each ligand adopt a distorted conformation (the dihedral angles range from 28.4° to 88.3°) within the non-coplanarity of the two phenyl rings. Besides, the amide moieties twist as well with respect to the pyridyl rings, and exhibit two roughly distinct arrangements. More specially, half of amide groups orients their C=O groups inward of the cavity, while the other half directs the C=O groups outward. These twist events thus result in the loss of the possible C4 symmetric of the structure.

|
Download:
|
| Fig. 3. The single crystal structures of 2. The solvent molecules, anions outside of cavity and disorders have been removed for clarity. | |
{kind=link}
We inferred that this structural torsion was mainly due to the requirement of the enthalpy gain in the compact structure. The CH-π and π-π interactions between the neighboring bridging ligands were evident in the crystal structure. Their proximities could also be verified by the NOESY experiment, in which the NOE cross peaks were obviously observed between the inner pyridine protons and the aniline aromatic protons (Fig. S2 in Supporting information). A total of eight hydrogen bonds (N-H…O, N-H…F) between the amide groups of the neighboring ligands as well as between the amide groups and the tetrafluoroborate anions were presented in the structure. All these interactions (hydrogen bonds, CH-π and π-π) arising from the twisting of the tertiary amine and amide moieties as well as the electrostatic interaction between the metals and the encapsulated anions contributed to stability of the interlocked cage.
The encapsulation of the BF4− anions were further supported by 19F NMR spectra, in which new peaks appeared except the free BF4− resonances (Fig. S3 in Supporting information). The competitive titration experiments showed that halide ions bound stronger to the cage than that of BF4−, while other anions such as PF6−, trifluoromethanesulfonate (OTf−) or bis(trifluoromethane)solfonimide (NTf2−) displayed little or none bind ability (Fig. S4 in Supporting information). Unexpectedly, addition of NO3− led to a decomposition of the cage. The control experiments showed that the interlocked cage could not be assembled when using NO3− or OTf− rather than BF4− as counter-anion. These results hint that the BF4− may be a template for assembly of the interlocked cage. The structural stability of 2 was examined by concentration- and temperature-variable NMR experiments subsequently. No evidence was found for decomposition of the complex either by diluting the solution to 0.01 mmol/L, or by raising temperature to 343 K, revealing its high thermodynamically stability.
In summary, we have synthesized a new interlocked cage, which is formed quantitatively through self-assembly of palladium(Ⅱ) ions and the aromatic amide ligands. Due to the flexibility of the ligand, the multiple supramolecular interactions are prone to be involved in stabilizing the structure. The twisting of amide moiety may also bring about the helical chirality of the system, which is currently being investigated in our laboratory and will be reported in due course.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21871101) and the Natural Science Foundation of Hubei Scientific Committee (Nos. 2017CFA036, 2019ACA125). We also thank the staffs from BL17B at Shanghai Synchrotron Radiation Facility, for assistance during crystal data collection.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.002.
| [1] |
C.M. Hong, R.G. Bergman, K.N. Raymond, et al., Acc. Chem. Res. 51 (2018) 2447-2455. DOI:10.1021/acs.accounts.8b00328 |
| [2] |
A.M. Nonat, L.J. Charbonnière, Coord. Chem. Rev. 409 (2020) 213192. DOI:10.1016/j.ccr.2020.213192 |
| [3] |
M. Pan, Z. Wei, Y. Xu, C.Y. Su, Prog. Chem. 29 (2017) 47-74. |
| [4] |
H. Jiang, Q. Li, G. Wang, Chin. J. Org. Chem. 38 (2018) 1065-1084. DOI:10.6023/cjoc201711013 |
| [5] |
F.J. Rizzuto, L.K.S. van Krbek, J.R. Nitschke, Nat. Rev. Chem. 3 (2019) 204-222. DOI:10.1038/s41570-019-0085-3 |
| [6] |
T.R. Cook, P.J. Stang, Chem. Rev. 115 (2015) 7001-7045. DOI:10.1021/cr5005666 |
| [7] |
N.B. Debata, D. Tripathy, D.K. Chand, Coord. Chem. Rev. 256 (2012) 1831-1945. DOI:10.1016/j.ccr.2012.04.001 |
| [8] |
S. Pullen, G.H. Clever, Acc. Chem. Res. 51 (2018) 3052-3064. DOI:10.1021/acs.accounts.8b00415 |
| [9] |
S. Saha, I. Regeni, G.H. Clever, Coord. Chem. Rev. 374 (2018) 1-14. DOI:10.1016/j.ccr.2018.06.010 |
| [10] |
L.J. Chen, H.B. Yang, M. Shionoya, Chem. Soc. Rev. 46 (2017) 2555-2576. DOI:10.1039/C7CS00173H |
| [11] |
K. Harris, D. Fujita, M. Fujita, Chem. Commun. 49 (2013) 6703-6712. DOI:10.1039/c3cc43191f |
| [12] |
M. Yoshizawa, K. Ono, K. Kumazawa, et al., J. Am. Chem. Soc. 127 (2005) 10800-10801. DOI:10.1021/ja053009f |
| [13] |
S. Zarra, D.M. Wood, D.A. Roberts, et al., Chem. Soc. Rev. 44 (2015) 419-432. DOI:10.1039/C4CS00165F |
| [14] |
M. Fujita, N. Fujita, K. Ogura, et al., Nature 400 (1999) 52-55. DOI:10.1038/21861 |
| [15] |
Y. Yamauchi, M. Yoshizawa, M. Fujita, J. Am. Chem. Soc. 130 (2008) 5832-5833. DOI:10.1021/ja077783+ |
| [16] |
T.K. Ronson, Y. Wang, K. Baldridge, et al., J. Am. Chem. Soc. 142 (2020) 10267-10272. DOI:10.1021/jacs.0c03349 |
| [17] |
A. Westcott, J. Fisher, L.P. Harding, et al., J. Am. Chem. Soc. 130 (2008) 2950-2951. DOI:10.1021/ja8002149 |
| [18] |
J.J. Henkelis, T.K. Ronson, L.P. Harding, et al., Chem. Commun. 47 (2011) 6560-6562. DOI:10.1039/c1cc10806a |
| [19] |
J. Heine, J. Schmedt auf der Günne, S. Dehnen, J. Am. Chem. Soc. 133 (2011) 10018-10021. DOI:10.1021/ja2030273 |
| [20] |
R. Sekiya, M. Fukuda, R. Kuroda, J. Am. Chem. Soc. 134 (2012) 10987-10997. DOI:10.1021/ja303634u |
| [21] |
S. Freye, J. Hey, A. Torras-Galán, et al., Angew. Chem. Int. Ed. 51 (2012) 2191-2194. DOI:10.1002/anie.201107184 |
| [22] |
M. Frank, M.D. Johnstone, G.H. Clever, Chem. Eur. J. 22 (2016) 14104-14125. DOI:10.1002/chem.201601752 |
| [23] |
Y.H. Li, J.J. Jiang, Y.Z. Fan, et al., Chem. Commun. 52 (2016) 8745-8748. DOI:10.1039/C6CC04420D |
| [24] |
R. Zhu, J. Lübben, B. Dittrich, et al., Angew. Chem. Int. Ed. 54 (2015) 2796-2800. DOI:10.1002/anie.201408068 |
| [25] |
R. Zhu, I. Regeni, J.J. Holstein, et al., Angew. Chem. Int. Ed. 57 (2018) 13652-13656. DOI:10.1002/anie.201806047 |
| [26] |
M. Fukuda, R. Sekiya, R. Kuroda, Angew. Chem. Int. Ed. 47 (2008) 706-710. DOI:10.1002/anie.200703162 |
| [27] |
D. Tripathy, A.K. Pal, G.S. Hanan, et al., Dalton Trans. 41 (2012) 11273-11275. DOI:10.1039/c2dt30937h |
| [28] |
M. Fukuda, R. Sekiya, R. Kuroda, Angew. Chem. Int. Ed. 47 (2008) 706-710. DOI:10.1002/anie.200703162 |
| [29] |
R. Sekiya, R. Kuroda, Chem. Commun. 47 (2011) 12346-12348. DOI:10.1039/c1cc14982b |
| [30] |
Q. Lin, L. Gao, B. Kauffmann, et al., Chem. Commun. 54 (2018) 13447-13450. DOI:10.1039/C8CC07932C |
| [31] |
B. Htan, D. Luo, C. Ma, et al., Cryst. Growth Des. 19 (2019) 2862-2868. DOI:10.1021/acs.cgd.9b00096 |