 2021, Vol. 32
2021, Vol. 32 


Directed by H-bonds between amides, polypeptides facilitate the formation of chiral secondary structures mainly comprising of α-helix and β-sheet. In contrast to the fact that β-sheet is more favorable in most peptide assemblies even for shortest dipeptide and individual amino acids [1, 2], helical structure formation requires more strict prerequisites on the numbers and types of amino acid residues. According to the Zimm-Bragg theory [3], equilibrium constant S (increasing one residue on the terminal of a helix) should be ≥ 1 when stable peptide helix forms. Such scenario only occurs in long peptide sequences. Zimm-Bragg theory predicts that peptides possessing less than 20 residues are unfavorable to form stable helices. For instance, for peptide with 13 residues, the odd for helix formation is only 8% [4]. In spite of Zimm-Bragg model, some exceptions have been found, mainly contributed by Gazit and coworks [5-7]. And a major advancement has been made recently revealing the supramolecular helical structure formation from a tripeptide sequence (Phe-Phe-Pro) in solid-state [6]. So far, it is the shortest peptide sequence for helical structure formation, and no α-helix like structures have been reported in self-assembled dipeptides, amino acids as well as their derivatives. Recently, Gazit [8] proposed a concept of supramolecular secondary structures by illustrating the formation of β-sheet motifs of zwitterionic coded amino acids, which inspired us to explore the possibility of superhelical structure formation in structural simple amino acid derivatives.
Among the amino acid derivatives, N-terminal aryl amino acids are promising species in constructing chiral self-assemblies [9-14]. Amide and carboxylic acid groups provide H-bonds for directing ordered molecular organization. In confined self-assemblies, aryl groups shall be appended with transferred chirality, illustrating chiroptical properties such as circularly polarized light absorption and emission. The absolute chirality of amino acids could be readily switched between L- and D- to study the enantiomer effect. In spite of these merits, aggregation of N-terminal aryl amino acids suffers from the crystallization-induced self-assembly, which hinders the 1 D growth into helical fibers [15, 16]. Consequently, it remains difficulties in recognizing the supramolecular chirality behaviours and properties such as handedness. Although circular dichroism (CD) and circularly polarized luminescence (CPL) spectrum could assist to identify the handedness or the occurrence of chirality, these techniques are limited to the aryl groups or luminophors [17]. To date, the structure-property relationship between supramolecular chirality and molecular topology is rather obscure. These challenges block the better understanding of the origin of supramolecular chirality in amino acid derivative aggregates as well as the rational design of chiroptical soft materials.
Through analyzing their solid state structures, here we provide a protocol to unveil the supramolecular helical structures of amino acid residue sequences and aryl groups respectively, as well as the insight into structure-interaction-chirality relationships. Typical 9-fluorenylmethoxycarbonyl (Fmoc) protected amino acids (L-) were selected as model building blocks. Eight Fmoc-amino acids with reported X-ray crystal structures which are freely available from the Cambridge Structural Database (CSD) are listed in Scheme 1. We searched α-helix like structures based on the principle of asymmetric H-bond induces asymmetric packing [6]. Unexpectedly, all the applied Fmoc-amino acids comprise exclusive asymmetric H-bonds, which are generated in three modalities including carboxylic acid (donor)→carboxylic acid (acceptor), amide→carboxylic acid and carboxylic acid→amide. H-bond induces the consecutive super-helical structures of amino acid residues and determines their handedness (the first two modalities give P-helix while the last one favors M-helix) as well as the helical pitch (for instance last modality give longest values). Accompanied with helical amino acid residues, fluorene segments stack into asymmetric supramolecular tilted helices, and in most cases (7 out of 8) demonstrate inverted chirality to amino acid sequences. This novel protocol reveals the role of carboxylic acid-involved H-bonds in determining super-helical structures in molecularly simple aryl amino acids. This work also provides solid evidence for the chirality inversion behaviors by altering carboxylic acid H-bonds utilizing carboxylic acid binders in aryl amino acid self-assemblies [10, 18-22]. It solves the curiosity about the diversified Cotton effects of achiral microsheets or ribbons from self-assembled aryl amino acids [13].

|
Download:
|
| Scheme 1. (a) The library of Fmoc-protected amino acids investigated in the present work. (b) Supramolecular helical chirality and tilted chirality found in Fmoc-amino acid packing arrays induced by distinct H-bonds. | |
{kind=link}
It is known that crystal lattice comprises of three-dimension extensions, and the protocol to determine the existence of helices refers to the Gazit's work [6] that supramolecular helices are induced by H-bonds. A representative case is demonstrated in Fig. 1: One Phe is capable of forming two set of intermolecular H-bonds including inter-amide and inter-carboxylic acid modality. However, the periodic inter-amide H-bonds are parallel to each other (Fig. 1b). Consequently, the continuous molecular junction exhibits neither asymmetrical orientation nor chiral sense. In contrast, the inter-carboxylic acid H-bond sequence is apparently asymmetrical with a Zig-Zag propensity (Fig. 1b). Highlighting the adjacent carboxylic groups by spacefill mode gives rise to obvious P-helix (Fig. 1c). The inter-carboxylic acid H-bond has higher ∠C-O-H angle of 147.3° (distance =1.80 Å). This property further shrinks the helical pitch to 4.9 Å. The supramolecular chirality has been transferred to the aryl groups covalently conjugated to amino acids. Due to the inconsecutive packing of aryl groups, the determination of chirality was by means of supramolecular tilted chirality developed by Miyata and coworkers [23]. Along the central axis (in this case, b direction), the angle θ which is defined as a clockwise horizontal rotation of tilted aryl planes around the central axis, indicates M- (0° < θ < 90°) or P-handedness (90° < θ < 180°). On top of this definition, the handedness of tilted chirality of aryl groups is identical to amino acid sequence, sharing a P-helicity.

|
Download:
|
| Fig. 1. Analysis of crystal packing structures of (a-d) Phe (CCDC: 1060776), (e) Asp (CCDC: 1532789) and (f) Ile (CCDC: 700540) to reveal how to determine the superhelical sense of amino acid residues and aromatic groups. (a) One Phe molecule could generate two set of H-bonds, which show parallel and asymmetrical orientations respectively (b). Highlighting the continuous molecular junction between asymmetrical H-bonds (c) and fluorene groups by spacefill mode give helical structures and supramolecular tilted chirality (d) respectively. (e, f) Asymmetric packing of Asp and Ile, respectively. Except for fluorene demonstration, green, red, blue and white stand for C, O, N and H atoms respectively. | |
{kind=link}
The first scenario found in crystals is the asymmetric H-bonds between adjacent carboxylic acids. It comprises of three building blocks which are Phe, Asp and Ile. Asp features an intramolecular H-bond between amide (donor) and ester (acceptor) group, allowing for the occurrence of intermolecular H-bonds between carboxylic acids. As shown in Fig. 1e, the H-bond with a distance of 1.84 Å and ∠C-O-H of 144.4° is stronger than that of amide-carboxylic acid found in Ala and Ser. Due to carboxylic acid groups function as both donor and acceptor, it gives rise to amino acid superhelix with relative high helical angle and low helical pitch. The helical pitch of Asp is merely 5.4 Å. P- and M-helicity was found in amino acid sequence and aryl group stacking respectively. Compared to Asp and Phe, Ile (Fig. 1f) packing lacks neither intramolecular nor inter-amide H-bonds. It is found that amide (donor) bonds to the O-H group (acceptor) of carboxylic acid. Consequently, the inter-carboxylic acid H-bond could be realized (1.77 Å and ∠C-O-H is 153.2°). The whole packing modality of Ile is almost identical to that of Asp (helical pitch 5.3 Å), and the helicity of amino acid and aryl groups is inverted from P to M.
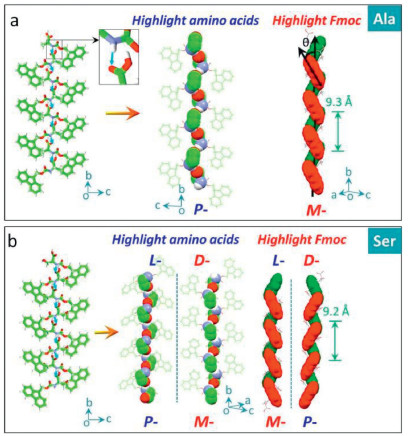
The second scenario is the asymmetric H-bonds between N-H/C=O from adjacent amide (donor) and carboxylic acid (acceptor) (Fig. 2). Ala and Ser fall into this circumstance. Specifically, water molecules are implanted into crystal lattice in an ordered manner, whereby one Ala or Ser molecule is bonded by three water molecules via H-bonds, and the H-bonds between N-H/C = O exclusively connect adjacent building blocks. The molecular packing leading by this H-bond is shown in Fig. 1. The spatially asymmetric H-bonds induce the asymmetrical packing of Ala and Ser. For Ala, the donor-acceptor distance of H-bond is 2.15 Å and ∠C-O-H is 131.7°. The molecular backbone linked by H-bonds is elucidated by spacefill mode, and the amino acid residues exhibit left-in/right-out spatial relationship, which is designated as right-handed P-helical chirality. The fluorene segment chirality was determined to be M-handedness, which is inverted compared to P-handed amino acid sequences. Due to the fixed configuration of Ala in solid states, the helical amino acid and fluorene moieties share the same helical pitch (9.3 Å). Ser shows almost identical H-bond to Ala, with a donor-acceptor distance of 2.08 Å and ∠C-O-H of 133.8°. Along b axis, P- and M-helicity were found in amino acid residues and aryl groups of Ser respectively. Helical pitch was determined as 9.2 Å. In order to verify the enantiomer-dependence of supramolecular chirality, the enantiomer of Ser was analyzed. Expectedly, D-Ser possesses mirror handedness to L-Ser, indicative of the supramolecular handedness by asymmetrical H-bonds could be tailored by the absolute configuration of amino acids.

|
Download:
|
| Fig. 2. Asymmetrical H-bonds between amide (donor) and carboxylic acid (acceptor). (a) Packing of Ala (CCDC: 1532788) along b axis enables the helical chirality of amino acid sequences and tilted chirality of aryl groups. (b) Asymmetric packing of Ser (CCDC: 1941464) and its enantiomer (CCDC: 1532790). | |
{kind=link}
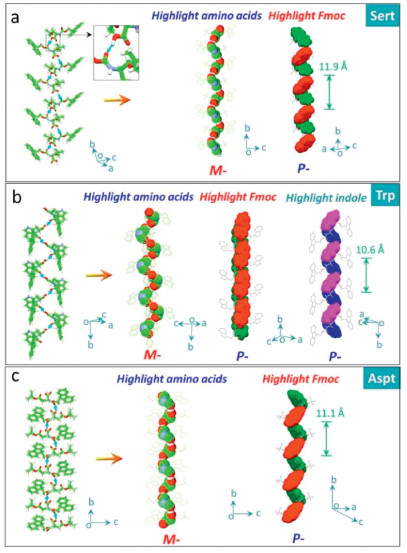
The third asymmetric H-bond we observed in solid state structures of applied Fmoc-amino acids is between the carboxylic acid and amide which behave as donor and acceptor respectively, in contrast to the second modality (Fig. 3). Trp, Sert and Asp accord with this feature, which are all conjugated with bulky groups with considerable steric hindrance (indole and t-butyl group). For Trp, the specific H-bond (distance and angle are 1.86 Å and 138.9°) is exclusive without the occurrence of other H-bond ascribed to the bulky effect of the indole group. This H-bond enables a left-handed M-helicity of amino acid sequences, in sharp contrast to the above P-helices. Although these Fmoc-amino acids share the same absolute point chirality (L-), the differential in supramolecular chirality suggests the key role of H-bond chirality in directing handedness. Uniquely, Trp contains two independent aryl groups, fluorene and indole, of which supramolecular handedness is determined as P-type. This modality generates high helical pitch up to 10.6 Å, almost two folds compared to the second scenario. Steric hindrance effect of t-butyl group in Sert (Fig. 3b) blocks the other H-bonds other than carboxylic acids (acceptor) and amide group (acceptor). The short distance of the H-bond (1.60 Å) implies the strong binding effect (bond angle = 137.7°). M- and P-handedness are found in amino acid residue and aryl groups respectively. A highest helical pitch was obtained (11.9 Å). Aspt follows the same binding modality due to the presence of t-butyl group (Fig. 3c). Like Asp, Aspt features intromolecular H-bond between ester group (acceptor) and amide group (donor), which barely interferes the asymmetrical H-bonds (distance and angle are 1.91 Å and 141.7°).

|
Download:
|
| Fig. 3. Asymmetrical H-bonds between carboxylic acids (acceptor) and amide group (acceptor). Amino acid residues and aryl groups are depicted in spacefilling mode respectively. (Trp CCDC: 1532791, Sert CCDC: 662774, Aspt CCDC: 754211). | |
{kind=link}
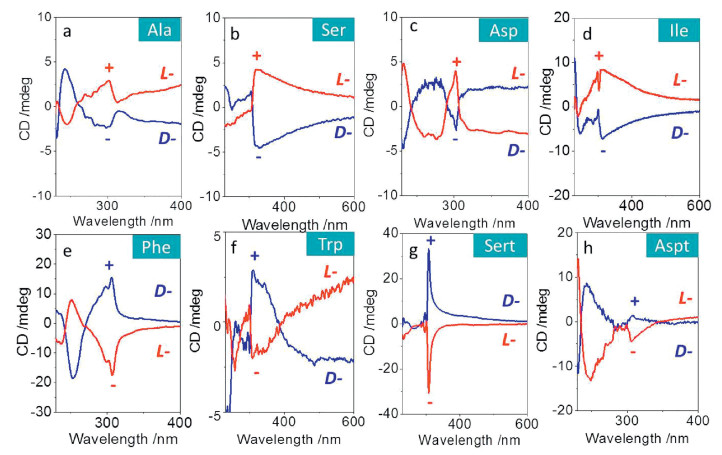
Through analyzing the solid-state structures, it can be concluded that the handedness of aryl groups (fluorene) comprises P- (Ala, Ser, Ile and Asp) and M-type (Phe, Trp, Sert and Aspt). Thus we considered wither the supramolecular handedness was associated with their chiroptical properties. To do so, Fmoc-amino acids were allowed to crystallize in acetone in the presence of minimal poor solvent water, and the resultant crystals were collected and fully grinded with KBr in order to eliminate the linear dichroism (LD) artifact effect. The absorption region of fluorene group is ranging from ca. 230 nm–300 nm (Fig. S1 in Supporting information), and the CD spectra show major Cotton effect peak at 300 nm (Fig. 4). The Cotton effects are enantiomer-resolved, whereby well-defined mirror spectra were obtained. For L-amino acids, Ala, Ser, Asp and Ile possess positive (+) Cotton effect at 300 nm, while Phe, Trp, Sert and Aspt demonstrate the negative (-) values. The chiroptic results are in good agreement with our conclusion of aryl handedness from crystal structures. This fine congruent relationship (M-helicity to positive and P-helicity to negative Cotton effects) would allow us to determine the asymmetric packing of aryl groups from CD spectra or predict the Cotton effects based on single crystal structures.

|
Download:
|
| Fig. 4. CD spectra of Fmoc-amino acids in crystalline states with L- and D-configuration. | |
{kind=link}
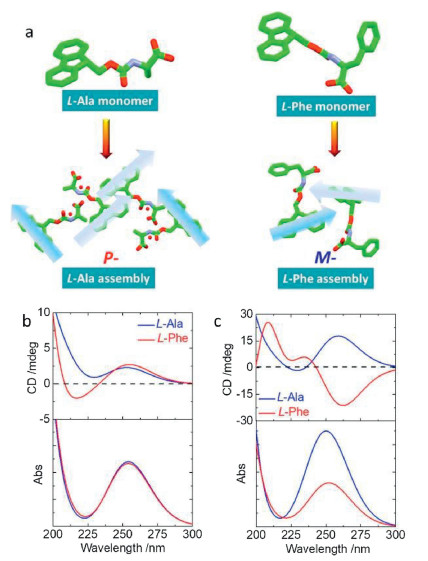
In order to gain further insight to the correlation between asymmetric packing in crystal and chiroptical responses, we further calculated electronic circular dichroism (ECD) spectra based on the time-dependent density functional theory (TDDFT) with the Gaussian 16 program. The configurations of Fmoc-amino acids were extracted directly from single-crystal structures (self-assembled structures were packing array in a unit cell), which were further optimized with the B3LYP/6-311G(d) basis set to obtain a low-energy conformation. Such optimization hardly altered the pristine geometries in both monomeric and self-assembled states (Fig. 5a). Based on the low-energy optimization, the rcam-B3LYP/6-311G(d) basis set and method were employed for TDDFT calculation to obtain the ECD spectra. For comparison, Phe and Ala (L-) that show distinct supramolecular tilted handedness with respect to fluorene segments were chosen (Fig. 5). In monomeric state, the absolute configurations give rise to absorption spectra centered at 255 nm, where both Ala and Phe feature a positive Cotton effect band (Fig. 5b). It indicates fluorene segments share the same asymmetric environment independent to amino acid residues. However, in self-assembled states, fluorene segments form chiral alignments with P- and M-handedness for Ala and Phe respectively. Significant variation has been observed for ECD spectra (Fig. 5c) after self-assembly. While Ala retains the positive Cotton effect at around 260 nm, Phe assembly demonstrates negative Cotton effect, which suggests fluorene segments are in opposite-handed packing modalities. Such results are in good agreement with our above assumption regarding the correlation between the observed tilted chirality and optical activities, which are determined by supramolecular tilted chirality in addition to the absolute chirality of amino acids. On top of the assumption, chiroptical properties could be readily speculated via single crystal or molecular dynamic simulated structures.

|
Download:
|
| Fig. 5. ECD spectra. (a) Optimized crystal structures of Ala and Phe in monomeric and aggregated states for ECD calculation. Calculated ECD spectra and corresponding absorption spectra of Ala and Phe in (b) monomeric state and (c) aggregated state. | |
{kind=link}
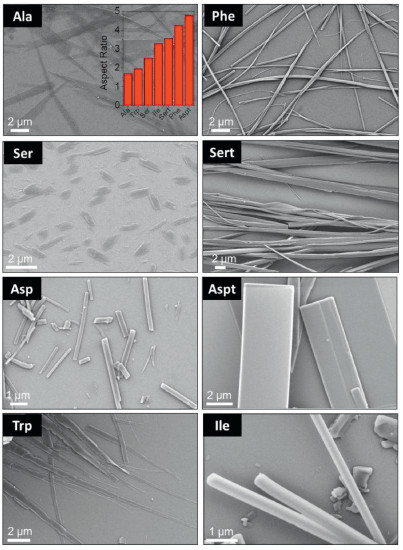
Crystal morphology was predicted based on the crystal structures by Materials Studio 7.0 program, and the corresponding largest aspect ratios are shown in the inset of Fig. 6. Asp failed to give aspect ratio value due to the existence of disordered structure in single crystal profiles. Most amino acids exhibit large aspect ratios high than 3.0, indicating their excellent capability in forming fibrous structures under kinetic conditions. Self-assembly of the Fmoc-amino acids was initiated by the typical nanoprecipitation protocol via solvent exchange. Concentrated hexafluoroisopropanol solutions were dispersed into bulky aqueous media to reach a specific concentration (2.5 mmol/L), when emulsion and precipitate phases were given. Scanning electron microscopy (SEM) was employed to characterize the morphologies (Fig. 6). Generally, most building units afford structures at microscale. Ala, Ser and Trp self-assemblies feature ribbon-like topology while Asp, Aspt and Ile possess rigid rod-like structures, following the crystallization-induced-assembly pathway. In contrast, Phe and Sert self-assembled into flexible fibrous structures with length up to 100 μm and width around 1 μm, in good consistent with their aspect ratio values.

|
Download:
|
| Fig. 6. SEM images of self-assemblies. Except for Ser (5 mmol/L); concentration was fixed at 2.5 mmol/L. Inset is the aspect ratio of different amino acids. | |
{kind=link}
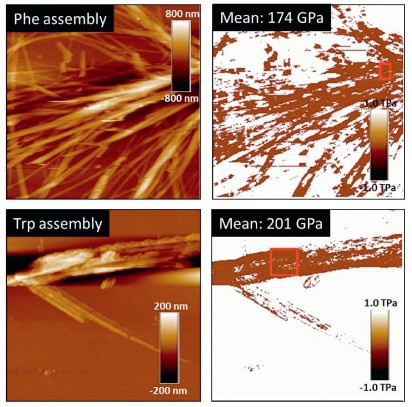
Crystalline self-assemblies of amino acids and peptides normally demonstrate quite large mechanical properties. For instance, Young's moduli of amino acid crystals could approach 100 GPa at selective facets [24]. We further evaluated Young's modulus of self-assemblies from some Fmoc-amino acids. Young's moduli were measured with a Bioscope Resolve atomic force microscopy (AFM). Deflection sensitivity was calibrated by ramp method using a standard sapphire sample. Spring constant k was calculated by Sader method, in which resonance frequency f0 and quality factor Q were calibrated using thermal tune method. Tip radius was calibrated using a standard Ti roughness sample, and indentation was calibrated by the sample in the qualify tip process. The Young's moduli were calculated by DMT models. Scan size is 0.3 Hz and samples/Line was 256, and Passion's ratio was assumed to be 0.3. Young's modulus mapping images are shown in Fig. 7, where morphologies are in agreement with SEM observations. Mean Young's moduli in selected areas were given in the insets, where Phe and Trp give a 174 and 201 GPa respectively. Such high values reflect rigid nature of the fibrous and ribbon structures, which are significantly higher than the unmodified amino acid crystals [21]. Although being appended with an aryl protected group at N-terminus, Fmoc amino acid self-assemblies show similar elastic mechanical properties to the rather rigid tri-peptide assembly reported by Gazit et al. [6].

|
Download:
|
| Fig. 7. AFM images of Phe (30×30 μm) and Trp (14.9×14.9 μm) self-assemblies as well as their corresponding Young's modulus mapping images. | |
{kind=link}
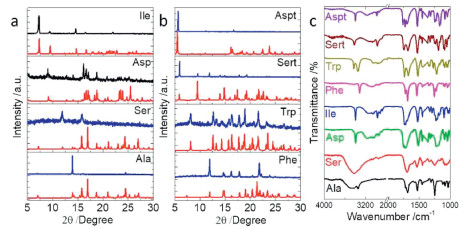
Self-assemblies were collected and subjected to powder X-ray diffraction (XRD) characterization to probe the preferential molecular extension (Figs. 8a and b). Compared to the simulated XRD pattern, Ala only demonstrates a very strong peak at 14°, which is assigned as (1, 0, 2) plane. Such behaviour indicates the pronounced exposure of this plane during kinetic assembly. Although Ser shares almost similar crystal packing arrays with Ala, its self-assembly clearly favours the exposure of (1, 0, 0) and (1, 1, 1) at 12° and 15.8° respectively. For the rest Fmoc-amino acids, almost identical XRD patterns were found compared to calculated results, in consistent to our speculation about the crystallization-induced self-assembly pathway, which apparently disregards the aqueous environment. Fourier transform infrared spectrum (FT-IR) was employed to probe the H-bonding formation in aqueous assemblies (Fig. 8c). A wide band at around 3400 cm−1 found in Ser and Ala samples is assigned as water residues, contributed to the entrapped water molecules in crystal structure. Stretching vibration of C = O at around 1700 cm−1 provides rich information with respect to H-bonding. Ala and Ser possess bands at 1692 cm−1 corresponding to the hydrated environments that carboxyl groups are fully involved to H-bonding. Phe, Ile and Asp show bands at 1731, 1727 and 1723 cm−1 respectively, consistent with dimeric carboxylic acid modality driven by inter-carboxylic acid H-bonding. In comparison, bands located at 1740, 1749 and 1766 cm−1 for Trp, Sert and Aspt respectively are contributed by the existing free carboxyl groups without being involved in any H-bonding. FT-IR spectra show well-defined congruent relationship with the carboxylic acid-involved H-bonding interactions in solid-states and in good agreement with our classifications on the helical superstructures based on carboxylic acid-involved H-bonding.

|
Download:
|
| Fig. 8. (a, b) XRD pattern comparison between calculated (red) and experimental (blue) results. (c) FT-IR spectra of different self-assemblies. | |
{kind=link}
In summary, we report a novel protocol to analysis helical sequences in N-terminal aromatic amino acids. In the applied Fmoc-amino acids, three asymmetric H-bond modalities were found, which play vital role in inducing the super-helical sequences of amino acid residues. Amide→carboxylic acid and carboxylic acid→carboxylic acid modality generates P-handed amino acid helices, while carboxylic acid→amide favours M-helix. In most cases except for Phe, the supramolecular tilted chirality of aryl groups shows inverted handedness to amino acids. Three distinct modalities also influence the helical pitch values. Our finding establishes a general tool to analyse the super-helical structures in solid state of aryl amino acids and unveil the critical role of carboxylic acid-involved H-bonds in directing the supramolecular chirality. Helical secondary structures induced tilted chiral aryl segments, which eventually determined their diversified chiroptical responses. Such structure-optical property relationship is firstly revealed in this work, which greatly facilitates the design criteria of chiroptical supramolecular materials.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work is supported by the Qilu Young Scholarship Funding of Shandong University. This works is also supported by the National Natural Science Foundation of China (Nos. 21872087, 21901145) and Natural Science Foundation of Jiangsu Province (No. BK20190209). We also acknowledge the financial support from Youth cross-scientific innovation group of Shandong University (No. 2020QNQT003). The authors acknowledge the assistance of Shandong University Structural Constituent and Physical Property Research Facilities. We thank Dr. Zhaozhen Cao from Shandong University for her kind assistance in chiroptical analysis.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.032.
| [1] |
X. Yan, P. Zhu, J. Li, Chem. Soc. Rev. 39 (2010) 1877-1890. DOI:10.1039/b915765b |
| [2] |
C.H. Görbitz, Crystallogr. Rev. 21 (2015) 160-212. DOI:10.1080/0889311X.2014.964229 |
| [3] |
B.H. Zimm, J.K. Bragg, J. Chem. Phys. 31 (1959) 526-535. DOI:10.1063/1.1730390 |
| [4] |
T.E. Creighton, Nature 326 (1987) 547-548. DOI:10.1038/326547a0 |
| [5] |
S. Mondal, L. Adler-Abramovich, A. Lampel, et al., Nat. Commun. 6 (2015) 8615. DOI:10.1038/ncomms9615 |
| [6] |
S. Bera, S. Mondal, B. Xue, et al., Nat. Mater. 18 (2019) 503-509. DOI:10.1038/s41563-019-0343-2 |
| [7] |
R. Parthasaratihy, S. Chaturvedi, K. Go, Proc. Natl. Acad. Sci. U. S. A. 87 (1990) 871-887. DOI:10.1073/pnas.87.3.871 |
| [8] |
S. Bera, S. Mondal, S. Rencus-Lazar, et al., Acc. Chem. Res. 51 (2018) 2187-2197. DOI:10.1021/acs.accounts.8b00131 |
| [9] |
S. Fleming, R.V. Ulijn, et al., Chem. Soc. Rev. 43 (2014) 8150. DOI:10.1039/C4CS00247D |
| [10] |
P. Xing, X. Chu, S. Li, et al., ChemPhysChem 15 (2014) 2377-2385. DOI:10.1002/cphc.201402018 |
| [11] |
P. Xing, X. Chu, M. Ma, et al., Chem. Asian J. 9 (2014) 3440-3450. DOI:10.1002/asia.201402645 |
| [12] |
P. Xing, P. Li, H. Chen, et al., ACS Nano 11 (2017) 4206-4216. DOI:10.1021/acsnano.7b01161 |
| [13] |
P. Xing, S.Z.F. Phua, X. Wei, et al., Adv. Mater. 30 (2018) 1805175. DOI:10.1002/adma.201805175 |
| [14] |
P. Xing, H. Chen, H. Xiang, et al., Adv. Mater. 30 (2018) 1705633. DOI:10.1002/adma.201705633 |
| [15] |
P. Xing, Y. Li, S. Xue, et al., J. Am. Chem. Soc. 141 (2019) 9946-9954. DOI:10.1021/jacs.9b03502 |
| [16] |
X. Yan, K. Zou, J. Cao, et al., Nat. Commun. 10 (2019) 3610. DOI:10.1038/s41467-019-11539-5 |
| [17] |
Y. Sang, J. Han, T. Zhao, et al., Adv. Mater. (2019) 1900110. |
| [18] |
G.F. Liu, L.Y. Zhu, W. Ji, et al., Angew. Chem. Int. Ed. 55 (2016) 2276. DOI:10.1002/anie.201511879 |
| [19] |
P. Xing, L. Bai, H. Chen, et al., ChemNanoMat 1 (2015) 517-527. DOI:10.1002/cnma.201500095 |
| [20] |
T. Xiao, L. Zhou, X.Q. Sun, et al., Chin. Chem. Lett. 31 (2020) 1-9. DOI:10.1016/j.cclet.2019.05.011 |
| [21] |
T. Xiao, L. Xu, J. Götz, et al., Mater. Chem. Front. 3 (2019) 2738-2745. DOI:10.1039/C9QM00595A |
| [22] |
T. Xiao, L. Xu, J. Wang, et al., Org. Chem. Front. 6 (2019) 936-941. DOI:10.1039/C9QO00089E |
| [23] |
A. Tanaka, I. Hisaki, N. Tohnai, et al., Chem. Asian J. 2 (2007) 230-238. DOI:10.1002/asia.200600332 |
| [24] |
I. Azuri, E. Meirzadeh, D. Ehre, et al., Angew. Chem. Int. Ed. 54 (2015) 13566-13570. DOI:10.1002/anie.201505813 |