 2021, Vol. 32
2021, Vol. 32 


Biological enzymes could regulate metabolic pathways and physiological processes in various organisms, making them an important class of biomarkers for disease diagnosis. Phosphatase is a large family of enzymes that could dephosphorylate in various biological materials, such as nucleic acids, proteins and alkaloids. It plays an important role in the regulation of cell signals and biological functions [1-3]. In the signal transduction pathway, phosphorylation is mainly used for signal initiation and amplification, while dephosphorylation is mainly related to signal termination. For example, PH domain leucine-rich repeat protein phosphatase (PHLPP) could specifically modulate the phosphorylation of human homolog to murine double minute 2 (HDM2) and glycogen synthase kinase 3α (GSK-3α) via dephosphorylating and inactivating Akt, leading to termination of Akt signaling which is related to cell growth and proliferation [4]. Based on the optimum pH for the activity, there is a subfamily of phosphatase enzymes that is widely distributed in cells, that is alkaline phosphatase (ALP) (EC 3.1.3.1).
ALP is a class of biological enzymes which are widely distributed in various organs, such as liver, bone, intestine, placenta, and other tissues [5]. It could remain maximum activity at pH around 8-11. As one of the most commonly tested diagnostic indicators in routine clinical analysis and test procedures, ALP has an important role in biochemical behaviors [6]. For example, ALP is a biochemical index in judging liver dysfunction clinically. The abnormal elevation of ALP is a common phenomenon in primary and secondary liver cancers [7]. Bone specific alkaline phosphatase (BALP) is a good marker of bone formation or neoplasm with bone metastases [8]. Some blood diseases such as leukemia usually have a close relationship with the decreased levels of ALP in serum [9]. Considering the level of phosphatase is closely related to many diseases, therefore, many methods have been explored to accurately detect phosphatase. Clinically, p-nitrophenyl phosphate (PNPP) is chosen to be the substrate of ALP to test their levels in the blood. With the hydrolysis by ALP, the PNPP could turn to p-nitrophenyl. The concentration of resultant p-nitrophenyl could be accurately quantified by the changes of absorption at 405 nm. Specially, a buffer solution of pH 11 is used for ALP detection. However, this invasive blood test is limited by poor compliance for patients. Most importantly, the detection of blood samples in vitro has a time lag. Thus, considering these aforementioned constraints of phosphate test in clinic, noninvasive methods which could detect phosphatase in real-time have attracted wide attention in the biomedical field.
So far, phosphatase detection methods are roughly divided into colorimetric method [10], fluorescence method [11, 12], chemiluminescence method [13, 14] and isotope labelling method [15]. Among these methods, the fluorescence method attracts great interests due to its simple operation, high sensitivity and low cost. Therefore, the development of fluorescence probes for visualization of phosphatase has become a hotspot. In this review, we summarize recent advances in fluorescence imaging of phosphatase. The fluorescence probes are classified according to their luminescence mechanisms, including intramolecular charge transfer (ICT), aggregation induced-emission (AIE), photoinduced electron transfer (PeT), excited state intramolecular proton transfer (ESIPT), rearrangement of conjugation system, fluorescence resonance energy transfer (FRET) and internal filtering effect (IFE). In the end, the challenges and prospects of phosphatase fluorescence probes are also discussed.
2. Design strategiesPredominant strategies for the design of fluorescent-based phosphatase probes can be divided into two categories. One is based on the fluorescence changes between the substrates and dephosphorylated products. This method usually suits for organic fluorescent probes which are composed of a fluorophore and a phosphate group which acts as a phosphatase recognition unit. Organic fluorescent probes generally have good biocompatibility and large half-peak width [13]. Through this method, the phosphatase activity can be well detected based on the fluorescence signal changes via two main ways, including ⅰ) turn on: the fluorescence signal at a special wavelength from "off" to "on"; ⅱ) ratiometric: the ratio of fluorescence signals at two special wavelengths. The illustration of this design principle is showed in Scheme 1. The other approach involves a substrate of phosphatase and a fluorescent nanomaterial. Specifically, the substrate or its catalytic product could influence the fluorescence property of nanomaterials, leading to the change of fluorescence signals when incubation with phosphatase. These substrates or products mainly contain PNPP [16], adenosine triphosphate (ATP) [17], ascorbic acid (AA) [18, 19], and so on. And this method is usually utilized to design inorganic probes. Inorganic probes possess many merits, such as easy preparation, high fluorescence quantum yield and large Stokes shift [16]. Considering many organic and inorganic phosphatase probes have been well designed and reported, we will introduce these fluorescent probes according to their luminescence mechanisms for better understanding.

|
Download:
|
| 1. Schematic illustration of ALP probes based on the dephosphorylation process. | |
2.1. ICT probes
ICT probes usually contain an electron donor and an electron acceptor. Considering the phosphate group can be cleaved by ALP, many researchers use the phosphate group as a recognition unit to influence the donor-π-acceptor balance of ICT probes [20]. Specifically, when the diagnostic reagent is in off state, the fluorescent group becomes in electron-deficient state, which leads to the entire system non-fluorescent. After enzymatic dephosphorylation, the phosphate group is removed from probes, resulting in the reconstruction of ICT. At this time, the activated probes can emit fluorescence signals at specific wavelengths.
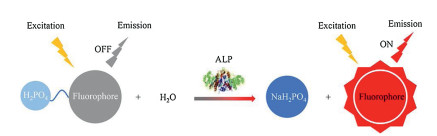
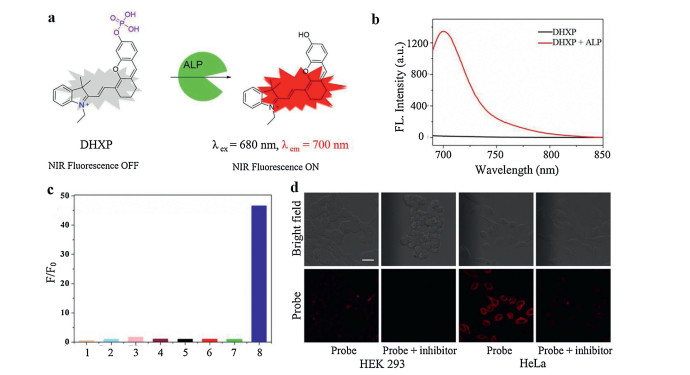
Many fluorophores such as hemicyanine and other compounds with strong ICT character have been widely explored for ALP detection. For example, Li et al. [12] utilized a hemicyanine derivative (CyOH) as the fluorophore to conjugate with the phosphate group. After the cleavage of phosphate group by phosphatase, 10-fold fluorescence enhancement could be achieved at 738 nm, promising sensitive phosphatase detection. Gwynne et al. [21] synthesized a 2-dicyanomethylene-3-cyano-4, 5, 5-trimethyl-2, 5-dihydrofuran (TCF)-based fluorescent probe (TCF-ALP) for the detection of phosphatase. Enzyme-induced hydrolysis of the phosphate group of TCF-ALP could turn this probe on with a 58-fold enhancement of fluorescence signal at 610 nm. Notably, this probe could respond to both acid phosphatase (ACP) and ALP at PBS (pH 7.4). Tan et al. [22] utilized a semi-cyanine fluorophore to conjugate with phosphate group, affording an ALP probe (DHXP) (Fig. 1a). In the presence of ALP, the activated probe expressed a 66-fold fluorescence enhancement at 700 nm (Fig. 1b). In striking contrast, as shown in Fig. 1c, negligible fluorescence signal could be detected when DHXP was incubated with ACP and phosphodiesterase (PDE), implying DHXP was a highly selective probe for ALP, therefore, other phosphatases would have little influence on the performance of this probe in biological applications. As shown in Fig. 1d, much stronger red fluorescence signal was exhibited in the HeLa cells (ALP-positive) than HEK 293 cells (ALP-negative) with the treatment of DHXP. Besides, the fluorescence signal of HeLa cells could be quenched by the ALP inhibitor sodium orthovanadate (Na3VO4). These results suggested DHXP could effectively detect the ALP in living cells.

|
Download:
|
| Fig. 1. (a) Mechanism of DHXP towards ALP detection. (b) Fluorescence intensity changes of DHXP when incubation with ALP. (c) The selectivity of DHXP (1: lysozyme; 2: esterase; 3: BSA; 4: avidin; 5: trypsin; 6: ACP; 7: PDE; 8: ALP). (d) Fluorescent imaging of DHXP in HEK 293 cells and HeLa cells. Reproduced with permission [22]. Copyright 2017, American Chemical Society. | |
It should be noted that inhibitors could help to clarify whether the resultant fluorescence signal is due to the dephosphorization of probes or other effects such as electrostatic interaction or hydrophobic interaction [23]. There are hundreds of inhibitors for phosphatases. For example, divalent cation-chelating agents like ethylene diamine tetraacetic acid (EDTA) have inhibition effects on ALP, while EDTA cannot inhibit the bioactivity of ACP. Conversely, fluoride ions such as sodium fluoride which has no effect on ALP could effectively inhibits the activity of ACP [24]. Other inhibitors such as phosphate and sodium orthovanadate could inhibit both ALP and ACP. In addition, there are four isoenzymes of ALP including intestinal, germinal, placental and tissue nonspecific ALP. Homoarginine can inhibit ALP isoenzymes except for placental ALP, and levamisole can inhibit ALP isoenzymes except for the intestinal and placental ones. In conclusion, an inhibitor can have inhibition effects on many enzymes, and an enzyme can be inhibited by many inhibitors. Thus, a smart choice should be made according to the aims. Recently, wang et al. [25] utilized a selective inhibitor (2, 5-dimethoxy-N-(quinolin-3-yl)benzenesulfonamide (DQB)) to ensure the intracellular function of fluorescent probes. Phosphatases distribute widely in the physical environment, such as tissue nonspecific ALP mainly distribute in extracellular environment and tyrosine phosphatase (PTP) mostly exist in membrane of mammalian cells [26]. By adding DQB that is a specific inhibitor for tissue nonspecific ALP, the fluorescence signal in extracellular environment could be largely decreased, promising enhanced accuracy for intracellular phosphatase visualization.
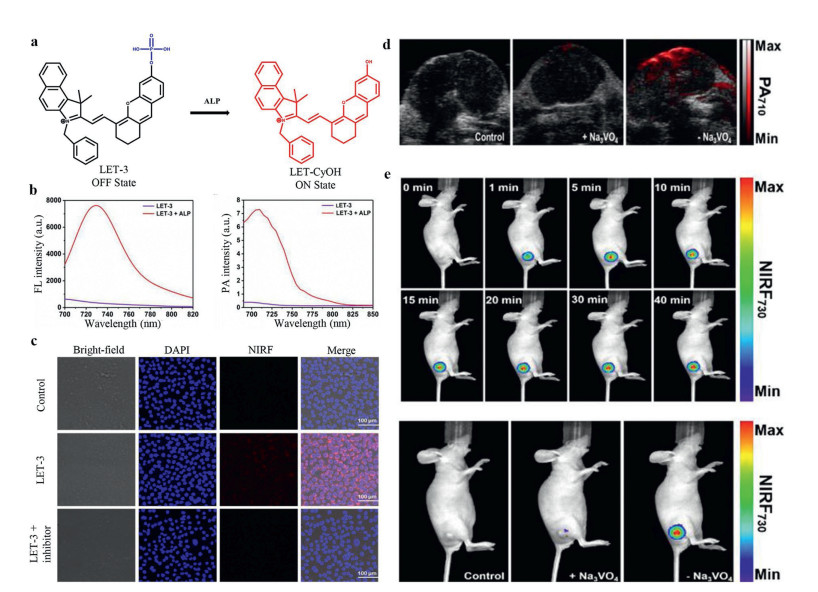
Compared with optical signals, acoustic signals have less scattering when signals pass through biological tissues, so the application of photoacoustic imaging (PA) can break the limit of imaging depth compared with traditional optical imaging. Recently, a fluorescence/photoacoustic dual-modality imaging probe (LET-3) based on phosphorylated cyanine had been developed for ALP detection (Fig. 2a) [27]. Upon incubation with ALP, the fluorescence intensity at 730 nm (NIRF730) and PA intensity at 710 nm (PA710) were enhanced to 23- and 27-fold, respectively (Fig. 2b). NIR fluorescence imaging was performed on the HeLa cells to validate the capability of LET-3 for ALP detection. Clear fluorescence signal was observed in the LET-3 incubated cells, and the fluorescence signal disappeared with additional adding an ALP inhibitor (Fig. 2c). Besides, the fluorescence/PA dual-modality imaging ability was also conducted in HeLa tumor-bearing nude mice. As shown in Figs. 2d and e, remarkable enhancement of PA enhancement at 710 nm and fluorescence signal at 730 nm were detected in tumor regions with the treatment of LET-3, while negligible PA and fluorescence were observed for the control group and group treated with Na3VO4. These results proved this dual-modality turn-on probe (LET-3) was suitable for ALP detection both in vitro and in vivo. In another similar work, Yao et al. [28] conjugated a mitochondria targeting group (triphenylphosphine) to the phosphorylated hemicyanine probe, affording an ALP probe for subcellular fraction sensing. While with addition of ALP, recovered fluorescence (λex = 735 nm) and PA signal at 710 nm could be observed. Except for excellent imaging function, this probe could eliminate the PC-3 tumors which were beared by BALB/c nude mice with irradiation (0.5 W/cm2) for 5 min due to the high photothermal conversion efficiency (η=35.9%).

|
Download:
|
| Fig. 2. (a) Fluorescence and photoacoustic dual mode detection of ALP activity by LET-3. (b) Fluorescence and photoacoustic signal when incubated with LET-3. (c) Fluorescence signals in Hela cells. (d) PA710 images of the group of control, the group received an intratumoral injection of Na3VO4 and LET-3, the group received an intratumoral injection of LET-3. (e) NIRF730 images of tumor-bearing mice treated with LET-3 at different time points, mice treated of saline (control) and mice treated with Na3VO4 then LET-3. Reproduced with permission [27]. Copyright 2019, American Chemical Society. | |
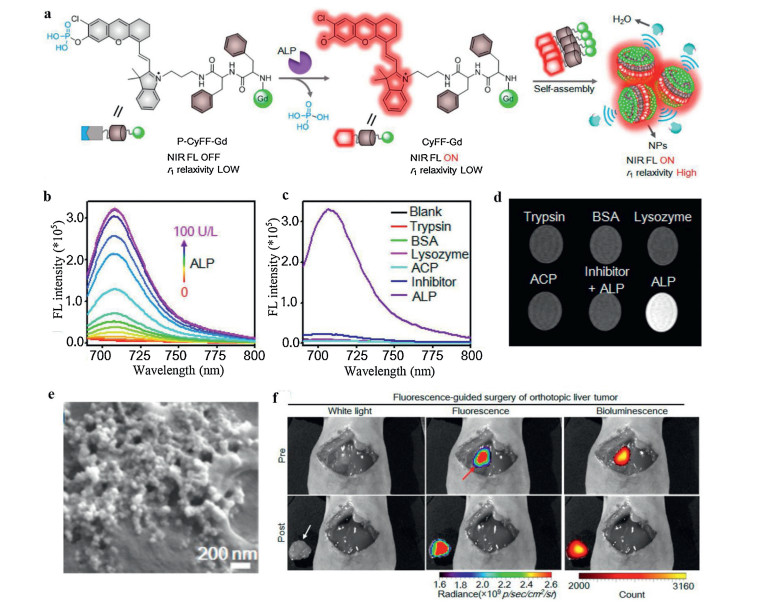
Considering most of the ALP is anchored on the surface of cell membrane, ALP-responsive in situ self-assembly process could be achieved by this ALP induced hydrophobicity changes [5, 29-31]. Instead of directly using prefabricated nanomaterials, this method utilizes small molecules that can easily pass through blood vessels and penetrate to target tissues. The ALP could remove the phosphate groups of probes, resulting in the changes of probe's hydrophobicity. In that case, the resultant amphiphilic compound could self-assembly in cell membrane to improve the cell uptake and retention. For example, Dong et al. [30] developed a new ALP probe incorporated a fluorescein-based precursor. With dephosphorylation by ALP, this probe had a sol-gel transition with fluorescence "turn-off" to yield aggregation caused quenching (ACQ). In another case, Yan et al. [32] utilized a hydrophobic dipeptide Phe-Phe linker to link phosphorylated hemicyanine and DOTA-Gd chelate (P-CyFF-Gd), promising multimodality imaging signals in one small molecule (Fig. 3a). DOTA-Gd is a clinically used magnetic resonance imaging (MRI) contrast agent. MRI can achieve excellent tissue penetration with high spatial resolution, which makes up for the deficiency of fluorescence imaging. Due to the hydrophilic phosphate group and DOTA-Gd groups, P-CyFF-Gd is super water-soluble with no fluorescence and low r1 relaxivity. As shown in Fig. 3b, with the hydrolysis of phosphate group by ALP, the newly generated phenolic hydroxyl group would enhance the ICT character of this probe, leading to more than 70-fold enhancement of fluorescence signal at 710 nm. Also, the selectivity of P-CyFF-Gd was tested by incubating P-CyFF-Gd with ALP and other representative enzymes. As shown in Figs. 3c and d, obvious enhancement of NIR fluorescence and MRI intensity were only showed in the P-CyFF-Gd group, implying this probe had a relatively high selectivity with negligible response to other enzymes. Specifically, with the leaving of hydrophilic phosphate group, this amphiphilic CyFF-Gd could self-assemble into nanoparticles, leading to restricted molecular rotation and prolonged tumbling time of DOTA-Gd chelate, thereby the r1 relaxivity was largely improved. Through ALP induced self-assembly, a new alternative for increasing sensitivity of MRI contrast agents could be achieved. As shown in Fig. 3e, uniform nanoparticles with diameter around 60 nm could be observed by cryo-SEM on the surface of HeLa cell membrane, implying the self-assembly behavior on the surface of cell membrane. Encouraged by these results, the in vivo imaging ability of P-CyFF-Gd was tested in orthotopic HepG2 liver tumors which express high level of ALP. Notably, luciferase transfected HepG2 cells was used to establish the liver tumors. As shown in Fig. 3f, compared with bioluminescence, remarkable fluorescence was showed in the liver after the treatment with P-CyFF-Gd, agreeing well with the bioluminescence imaging region of tumor, thus it could report accurate delineation of tumor margins for surgical resection.

|
Download:
|
| Fig. 3. (a) The self-assembly mechanism of P-CyFF-Gd probe. (b) Fluorescence intensity when incubated with ALP with different concentrations. (c) The fluorescent selectivity of P-CyFF-Gd for ALP against other enzymes. (d) T1-weighted MR images of P-CyFF-Gd incubated with ALP and other enzymes in Tris buffer (pH 8.0). (e) The cyto-SEM image of self-assembled nanoparticles. (f) Imaging guided surgical resection of orthotopic HepG2/Luc liver tumor after directly spraying P-CyFF-Gd on liver. Reproduced with permission [32]. Copyright 2019, American Chemical Society. | |
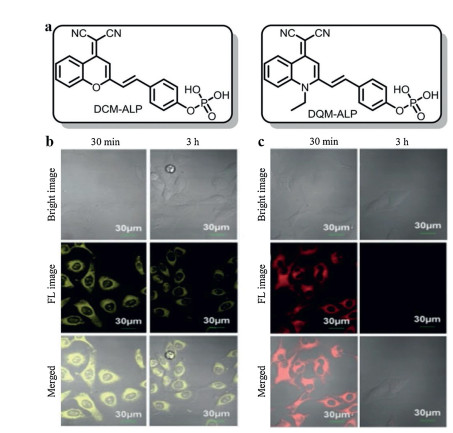
The phosphate group has a great influence on the hydrophilic-lipophilic properties of ALP probes. Thus, this attribute could be well utilized to design AIE probes. Typically, AIE probes are nonemissive in free forms but emit strong fluorescence in aggregate or solid forms. These solid forms are conducive to precise positioning by stick firmly in situ, thereby AIE probes could capture in situ information about relevant enzymes in the cells [33, 34]. Importantly, bioimaging with AIE probes tends to remain longer-term and higher-fidelity than that with ACQ probes. For example, Li et al. [35] utilized phosphate moiety as the phosphatase recognition group to conjugate with benzopyran and quinoline-malononitrile (QM) derivatives, respectively, affording an ACQ probe (DCM-ALP) and an AIE probe (DQM-ALP) (Figs. 4a). As shown in Figs. 4b and c, when incubation with HeLa cells for 3 h, the fluorescence signal of DCM-ALP almost totally vanished, while that of DQM-ALP still remain 66.98% of the original value. That was because the relatively larger size of aggregation could reduce the rate of diffusion or exocytosis.

|
Download:
|
| Fig. 4. (a) The structural formulas of DQM-ALP and DCM-ALP. (b) Fluorescence images of HeLa cells treated with DQM-ALP at 30 min and 3 h. (c) Fluorescence images of HeLa cells treated with DCM-ALP at 30 min and 3 h. Reproduced with permission [35]. Copyright 2020, John Wiley and Sons. | |
Beneficial from this unique property of AIE probes, many excellent ALP probes have been explored based on hexaphenylsilole (HPS), tetraphenylethylene (TPE) and QM derivatives, which are characterized as AIEgens with low signal-to-noise ratios. For example, an AIE probe based on TPE derivative, designated as TPE-PO3H2, was developed by Gu et al. [33]. Because of –PO3H2 group, the TPE-PO3H2 held a good solubility but poor fluorescence in aqueous solution. ALP-catalyzed hydrolysis of TPE-PO3H2 could induce the production of TPE. TPE with low water solubility could form aggregations and exhibit intense fluorescence with the emission peak at 480 nm. Besides, the fluorescence intensity had a good linear relationship with the concentration of ALP.
The turn-on and turn-off methods can merely detect the single change (e.g., increase or decrease) of fluorescence intensity, and these methods are vulnerable since many factors may influence its accuracy, such as pH and polarity of the media, light source intensity, photobleaching fluctuations [36]. In order to figure out this problem, Song et al. [37] developed a ratiometric AIE probe by conjugating chalcone derivative (HAC) with phosphate group for ALP visualization in living cells. Ratiometric probes can overcome the aforementioned drawbacks because it establishes a correction for environmental effects such as the variation of probe's concentrations [38]. This phosphorylated HAC (HCAP) was water-soluble and could yield greenish-yellow fluorescence in water. As shown in Fig. 5a, with the biocatalyst of ALP, water-insoluble HCA was gradually produced and thus activated AIE effect. Specifically, 2′-hydroxychalcone and its derivatives are recognized as fluorophores with ESIPT process [39]. In the crystallizing state, there existed strong intramolecular hydrogen bondings in HCA, which facilitated ESIPT process through the transformation from enol form to keto form, promising HCA with high fluorescence quantum yield and large Stokes shift. Because of the AIE and ESIPT, the aggregation of HCA had a strong fluorescence signal at 641 nm. Therefore, with the reaction with ALP, two non-interfering readings at 539 nm (HCAP) and 641 nm (HCA) could be yielded to form perfect ratiometric ALP probe (Fig. 5b). Besides, there was a good linear relationship between I641/I539 and ALP activity, suggesting HCAP could be used for accurate detection of ALP activity (Fig. 5c). HCAP probe was further applied for ALP mapping in living cells. As shown in Fig. 5d, by adding HCAP, a remarkable orange fluorescence signal of HCA was showed in HeLa cells. In contrast, the cells with no HCAP and cells treated with levamisole showed no fluorescence signal of HCA, implying HCAP had a well response in living cells. Importantly, the fluorescence of HCAP in cytoplasm did not increase with the addition of inhibitor, implying HCAP was reacted with the ALP secreted by cells, and resultant HCA then entered the cells and showed orange fluorescence.

|
Download:
|
| Fig. 5. (a) The mechanism of HCAP for ALP activity detection assay. (b) PL spectra of HCAP upon addition of ALP at different time points. (c) The linear relationship between the I641/I539 and ALP activity. (d) Fluorescence images of HeLa cells. (A, C, E) Bright field images corresponding to the fluorescence images with (B) no HCAP, (D) HCAP, (F) HCAP and levamisole. Reproduced with permission [37]. Copyright 2014, American Chemical Society. | |
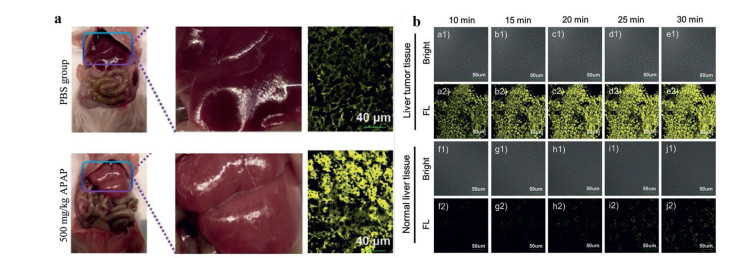
As mentioned above, an AIE probe DQM-ALP was synthesized by using QM derivative as the fluorophore. With the activation by ALP, the resultant aggregation could emit strong fluorescence at 550 nm which had a good linear relationship with the concentration of ALP. Considering the elevated level or bioactivity of ALP were recognized as a biochemical index in judging liver dysfunction clinically, the DQM-ALP was tested in acetaminophen (APAP)-induced acute liver injury model (ALI). As shown in Fig. 6a, compared with that of normal mice, obvious stronger fluorescence signal could be detected in liver tissues of mice with ALI after incubation with DQM-ALP, implying the increased concentration of ALP in the injured liver, and that was favorable for ALI predictions by fluorescence diagnosis. Furthermore, DQM-ALP was incubated with liver tumor tissues and normal liver tissues, respectively. As shown in Fig. 6b, obvious granular fluorescent spots which increased with prolonged incubation were exhibited in the tumor tissues incubated with probes. However, for the normal tissues incubated with DQM-ALP, almost no fluorescence signal could be observed after 10 min and only weak fluorescence signal was showed after 30 min, implying DQM-ALP could well distinguish the tumor cells from normal cells. This attribute is of vital importance because accurate differentiation of normal tissues and tumor tissues is beneficial for tumor resection and reduced cutting which have a profound significance for clinical surgery [35].

|
Download:
|
| Fig. 6. (a) Imaging of ALP regulation in the mice treated with PBS or APAP for 12 h. (b) Fluorescence images of 10 mm hepatocellular carcinoma tissue (a–e) and normal liver tissue (f–j) when treated with DQM-ALP. Reproduced with permission [35]. Copyright 2020, John Wiley and Sons. | |
In the first excited singlet state, the hydroxyl groups of probes are much more acidic and the hydrogen bond acceptors become much more basic compared with that in the ground state, promising significantly easier proton transfer from donor groups to acceptor groups by intermolecular or intramolecular hydrogen bond, and that is ESIPT. The most commonly used fluorophores for ESIPT are 2-(2′-hydroxyphenyl)-1, 3-benzoxazole (HBO), 2-(2′-hydroxyphenyl)benzothiazole (HBT), 2-(2′-hydroxy-3′-methoxyphenyl)benzothiazole (HMBT) and so on [40]. Modification of the hydroxy group of fluorophores with recognition unit could block ESIPT, leading to exclusively enol-like emission. After incubation with analytes, the restored hydroxyl groups could restart the ESIPT, promising a fluorescence change. For example, phosphate moiety was conjugated with HBT to yield a probe (HBT-P). This probe exhibited a fluorescent emission peak at 384 nm, which was a classic enol-like emission. When this probe was treated with phosphatases, a new emission peak occurred at 535 nm, which was characteristic of keto tautomer emission of HBT. Most importantly, HBT-P is the first ESIPT probe which could selectively detect protein tyrosine phosphatase-MKP-6 among the large protein PTP family. Unfortunately, the reason for this extraordinary selectivity of HBT-P has not been figured out [41]. Usually, ESIPT fluorophores in the electronic ground state typically exist in an enol form, while keto-enol tautomerism could happen in the excited state. Due to enol and keto forms of fluorophore exhibit different peaks of emissions, thus ESIPT probes with extraordinarily large Stokes shift could be used to establish ratiometric fluorescent probes [38]. Recently, Fan et al. [38] utilized ESIPT mechanism to establish a phosphatase probe. Specifically, phosphate moiety was conjugated with the 2-(2′-hydroxyphenyl)benzimidazole (HPBI) to mask the hydroxyl groups, leading to a single emission at 363 nm because of the blockage of ESIPT. With the addition of ALP, the ESIPT of HPBI was restored due to the resultant hydroxyl group, leading to a decrease of the fluorescence peak at 363 nm and an increase of a new emission band at 430 nm. Thereby, a ratiometric fluorescent phosphatase probe was established with a detection limit as low as 0.0013 U/mL.
Two interesting [42] were explored by Jia and his coworkers [43]. Specifically, nucleotides adenosine monophosphate (AMP) and guanosine monophosphate (GMP) were functioned to the HBT-P, respectively (Fig. 7a). These probes only displayed enol-like emission because the hydroxyl groups of HBT were masked. However, with the addition of ALP, an obvious increase in fluorescence signal at 512 nm was occurred due to the emission of the keto form. Importantly, both probes exhibited a highly selectivity for ALP against other types of phosphatases such as ACP and phosphodiesterase in PBS buffer (pH 7.4) (Figs. 7b and c). As mentioned before, HBT-P did not respond to ALP, implying the AMP and GMP played an important role in the selectivity of ALP. When incubation this probe with HeLa cells, an obvious fluorescence signal could be observed, while that of HT-29 cells (ALP negative) was negligible, implying these probes could be used to detect ALP in cellular environment. Specially, this probe was appiled in a zebrafish model. As shown in Fig. 7d, an obvious increase in fluorescence signal was observed over its belly/fins with the prolongation of time.

|
Download:
|
| Fig. 7. (a) The AMP (R1 = NH2, R2 = H)/GMP (R1 = O, R2 = NH2)-benzothiazole-based ALP probe. (b) Fluorescence signals at 512 nm during a real-time turn-on assay of ATP-based probe. (c) Fluorescence signals at 512 nm during a real-time turn-on assay of GMP-based probe. (d) ALP probe applied in zebrafish models for 2 min (1), 10 min (2), and 30 min (3). Reproduced with permission [43]. Copyright 2015, John Wiley and Sons. | |
It is worth mentioning that huge interests have been risen for combining ESIPT with other luminescence mechanisms. AIE is a promising complement to ESIPT due to the aggregates with a high signal-to-noise ratio could overcome the environmentally sensitive nature of ESIPT. The combination of ESIPT with FRET could yield a probe with longer wavelength absorption and emission, which is conducive to imaging in vivo [40]. Other mechanisms which could combine the favorable attributes of each other should be thoroughly researched in the future.
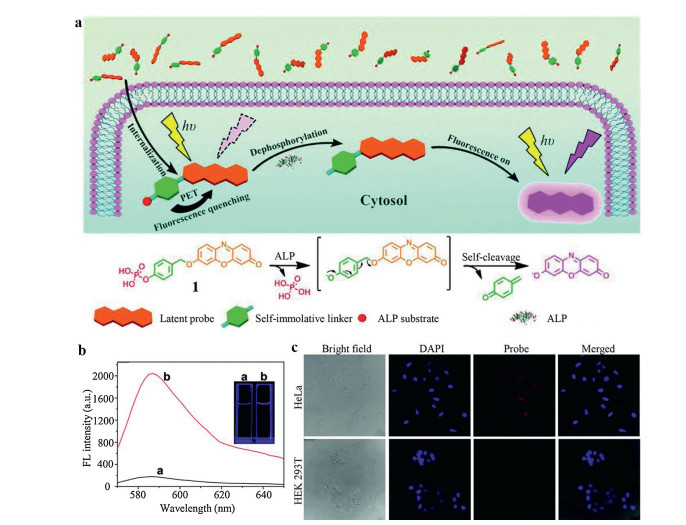
2.4. PeT probesThe PeT system is composed of an acceptor part, a fluorophore and a spacer [44, 45]. When the probe molecule is irradiated by light, the fluorophore does not or only emits weak fluorescence due to the excited electron is transferred from the donor to acceptor [44]. When the probe is reacted with the analyte, the PeT effect is blocked, and the fluorescence signal will be restored. To prove this concept, Zhang et al. [46] utilized a reactive p-hydroxybenzyl group as the spacer to link phosphate moiety with resorufin, termed as RhP. Upon interaction with ALP, the dephosphorylation reaction could be initiated and followed with a self-immolative reaction via 1, 6-elimination of spacer (Fig. 8a). In this case, the resorufin could be released with a strong emission at 585 nm (Fig. 8b). This probe was further tested in HeLa cells and HEK 293T cells. As expected, incubation with RhP, HeLa cells showed strong red fluorescence signal upon photo irradiation. Conversely, no obvious fluorescence signal could be observed in HEK 293T cells under the same condition (Fig. 8c). Actually, the fluorescence intensity in HeLa cells was 16-fold higher than that of stained HEK 293T cells by flow cytometry detection, implying the good response of this probe towards ALP. It is worth mentioning that resorufin is a classic ICT fluorophore that should be easily masked by suppressing the donor quality of the phenolic hydroxyl. However, compared with PeT probe, the resorufin-7-O-phosphate responded to ALP at a much reduced rate. Specifically, the Kcat/KM of RhP was 2.5-fold higher than that of resorufin-7-O-phosphate, implying the self-immolative linker could lead to a higher enzymatic efficiency. Considering the RhP had a smaller KM, the improved enzymatic efficiency might due to the introduction of a linker between resorufin and the phosphate group which could reduce the steric hindrance and enhance accessibility towards ALP.

|
Download:
|
| Fig. 8. (a) Illustration of the PeT probe for ALP enzymatic detection. (b) Fluorescence intensity of probe with or without ALP. (c) Fluorescence images of HeLa cells and HEK 293T cells. Reproduced with permission [46]. Copyright 2015, Royal Society of Chemistry. | |
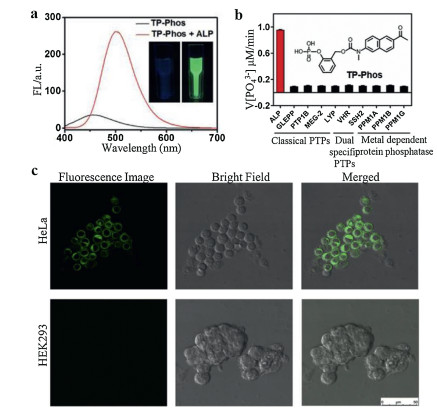
Compared with one-photon fluorescent probes, two-photon (TP) fluorescent probes could yield higher resolution and lower tissue autofluorescence during bio-imaging and therefore is very useful for visualization of deep tissues. Zhang et al. [47] utilized a self-cleavable linker to bridge a TP fluorophore with phosphate group to detect ALP in living cells, this probe was termed as TP-Phos. Similarly, with the hydrolysis of phosphate group by ALP, this probe could yield strong green fluorescence with an emission at 500 nm (Fig. 9a). Considering the phenyl phosphate group is the specific recognition site of PTP, thus the selectivity of TP-Phos towards ALP versus other classical PTPs was well tested. The dephosphorylation rate of TP-Phos with addition of ALP was 10-fold faster than that of PTPs (Fig. 9b). In contrast, 6, 8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) which is a commercially available ALP fluorescent probe showed poor selectivity between ALP and PTP. The high selectivity of the TP-Phos could be attributed to the ortho-functionalized phenyl phosphate group, which increased the steric hindrance of the probe into the deep and narrow pocket of classical PTPs [48]. Therefore, TP-Phos had good selectivity for ALP. Still, the imaging ability of TP-Phos was tested in HeLa cells and HEK 293 cells. Under excitation, the HeLa cells treated with the TP-Phos showed remarkable fluorescence signals while negligible fluorescence signal was showed in HEK 293 cells, implying the probe had an ALP detection ability in intracellular environment (Fig. 9c).

|
Download:
|
| Fig. 9. (a) Fluorescence intensity of TP-Phos with or without ALP in Tris-HCl buffer. (b) Selectivity studies of TP-Phos with ALP and various phosphatases. (c) Two-photon confocal images of HeLa cells and HEK 293 cells when incubated with TP-Phos for 10 min. Reproduced with permission [47]. Copyright 2017, Elsevier. | |
The luminescence mechanisms introduced above largely depend on the energy levels of molecular orbitals, which are hard to determine accurately, leading to poor prediction of the performance of the designed probes. In contrast, directly changing the conjugation system of probe which usually produces obvious changes of fluorescence signals is easier to predict [49]. Specifically, there are two ways to establish probes based on the change of π-conjugated system: nucleophilic addition reaction and ring-closing reaction. And for fluorophores, fluorescein, coumarin, spiropyran, rhodamine, cyanine and their derivatives could be used to construct this conjugation system via phosphorylation of hydroxyls.
Recently, Podder et al. [50] utilized a phosphate group to conjugate with fluorescein derivatives to establish a selective ALP and ACP probe, termed as LP1 (Fig. 10a). Specifically, the central quaternary carbon destroyed the π-conjugation of the xanthene moiety, leading to negligible fluorescence signal of LP1 in the visible region. After phosphatase hydrolysis, the large conjugation system of xanthene moiety was restored by intramolecular electronic rearrangement, promising strong fluorescence at 532 nm. Interestingly, LP1 had different responses at different pH. LP1 exhibited rapid increment of fluorescence signal with variable concentrations of ALP or ACP at pH 4.5. While at pH 7.4, only ALP could hydrolyze LP1 and yield strong fluorescence. In that case, LP1 can discriminate ALP from ACP in organelles with a higher pH. This probe was utilized to track the relative phosphatase expression in cancer cells (HeLa cells) and normal cells (NIH-3T3 cells). Dose-dependent fluorescence signals were both found at HeLa cells (Fig. 10b) and NIH-3T3 cells (Fig. 10c). However, obvious stronger fluorescence intensity was shown in HeLa cells treated with LP1 than that of NIH-3T3 cells, implying LP1 could discriminate cancer cells from healthy cells via tracking the expression of phosphatase.

|
Download:
|
| Fig. 10. (a) The mechanism of LP1 for phosphatase detection. (b) Dose-dependent fluorescent imaging of HeLa cells treated with LP1. (c) Dose-dependent fluorescent imaging of NIH-3T3 cells treated with LP1. Reproduced with permission [50]. Copyright 2018, the Royal Society of Chemistry. | |
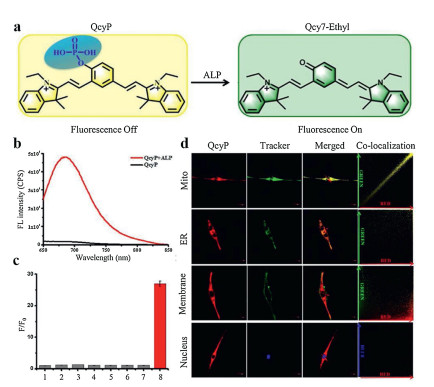
In another case, Ou et al. [1] also utilized fluorescein derivatives to detect ALP. Specially, a piperazine bridge was utilized to link phosphorylated rhodol and azido group modified coumarin, promising a single molecular with two reactive sites which could respond to phosphatase and H2S respectively. This probe was termed as N3-CR-PO4 (Fig. 11a). After reaction with H2S, the azido group could be reduced to amino group, affording a 13-fold enhancement of blue fluorescence signal at 445 nm (λex = 360 nm). Similarly, when incubation with ALP, it showed a recovery of fluorescence signal of rhodol at 545 nm with 23 times enhancement (λex = 510 nm). Besides, there was no spectral interference due to the large gap between these two emissive peaks which guaranteed the simultaneously accurate detection of phosphatase and H2S. Importantly, there existed a FRET effect between rhodol (absorber) and coumarin (donor). Thus, when coexisted with H2S and ALP, FRET between resultant rhodol and coumarin could be triggered and yield a green fluorescence signal at 545 nm (λex = 360 nm) (Fig. 11b). Therefore, this probe with three-channel imaging could be used to separately detect H2S, phosphatase and study the relationship between phosphatase activity and H2S level. This probe was further incubated with HeLa cells with addition of H2S scavenger (horbol 12-myristate 13-acetate (PMA)) or H2S to study the correlation between phosphatase activity and H2S level. Interestingly, both increase and decrease of H2S levels could greatly inhibit the bioactivity of phosphatase, implying even a slight deviation of the H 2S level would cause a sharp decrease in the bioactivity of phosphatase (Fig. 11c). This novel probe is simple but with powerful features, which is very helpful for in-depth research of molecule biology, especially the signaling transduction of enzymes. However, it should be noted that the region of yellow fluorescence seemed a little bit larger than that of blue fluorescence from the three-channel imaging. Considering the FRET only occurred when there existed both H2S and ALP, thus the ideal yellow fluorescence should only be showed in the intersection of the blue and green fluorescence. Therefore, probes with multiple channels which could capture in situ information in cellular environment should be explored in the future. Apart from fluorescein derivatives, cyanine is another favored group of synthetic π-conjugated dyes featured by a polymethine chain which has a charged and symmetric structure with a peripheral electron acceptor or donor group. Cyanines are highly valued for their easy modification and tunable properties. Appropriately tailoring the skeletons of cyanines could change their conjugation systems [51]. For example, Gao et al. [52] utilized heptamethine cyanine quinone derivative as fluorophore to conjugate with phosphate group to detect ALP, and this probe was abbreviated as QcyP (Fig. 12a). With the addition of ALP for 30 min, this probe offered strong fluorescence signals with a maximum wavelength of 685 nm (Fig. 12b). The mechanism for this turn-on probe was based on the conjugation system change. Specifically, by introducing a phosphate group to the central phenolic hydroxyl group, two positive charges were showed in the resultant probe, leading to the breakage of this π-conjugated system. In that case, no or only weak fluorescence signal could be detected. Upon incubation with ALP, the phosphate group was removed immediately from the cyanine skeleton, promising restored π-conjugated system via electron rearrangement, thereby, this probe was turned on accordingly. Besides, when incubation QcyP with ACP, phosphodiesterase and other hydrolases at physiological conditions (pH 7.4), there was no fluorescence at 685 nm, implying ACP and phosphodiesterase would not cause any interferences in the working solution (Fig. 12c). Importantly, this probe was used to colocalized the subcellular organelles in cells. Commercial dyes which could selectively stain mitochondria, endoplasmic reticulum (ER), cell membrane, and nucleus were utilized to trace the biodistribution of QcyP. As shown in Fig. 12d, overwhelming fluorescence signal of QcyP was showed in mitochondria with Pearson's colocalization coefficient as 0.91, while negligible fluorescence signal was occurred in ER, cell membrane and nucleus, implying QcyP predominantly located at mitochondria and could successfully respond to ALP in mitochondria. That was partly due to the cyanine skeleton had the instinct capability of selectively locating mitochondria. Regrettably, lysosome which also had a high level of ALP and ACP was not tested in this work. Subcellular colocalization is a meaningful work because it is crucial for understanding the interaction between the probe and analytes in intracellular environment. In another case, Zhang et al. [23] utilized phosphate group to mask the meso hydroxyl group of cyanine skeleton (Cy-O) to achieve a ratiometric probe (Cy-OP)via modulating the π-conjugated system of the cyanines. Unlike QcyP, the conjugation system of Cy-OP was not broken by introducing phosphate group, promising a fluorescence signal at 766 nm. With the departure of phosphate esters, Cy-OP would be transformed into the conjugation structure of Cy-O via electron rearrangement, leading to a blue shift of fluorescence signal with the maximum wavelength of 616 nm. Thus, a ratiometric probe with large Stokes shift could be well established. It was worth mentioning that Cy-OP had little response when the pH was below 6.5 due to the dephosphorylated product tended to be protonated at acid environment, leading to failed electron rearrangement. Similarly, as-prepared Cy-OP also tended to colocalized at mitochondria.

|
Download:
|
| Fig. 11. The mechanism of the fluorescence response towards H2S and phosphatase. (b) Fluorescent spectra of mixing 10 mmol/L N3-CR-PO4 and ALP with the addition of 50 μmol/L H2S by the excitation of 360 nm. (c) The influence of different levels of H2S on the activity of phosphatase in live cells (CH1: H2S detection; CH2: phosphatase detection; CH3: the fluorescence signal of FRET probe). Reproduced with permission [1]. Copyright 2019, John Wiley and Sons. | |

|
Download:
|
| Fig. 12. (a) The detection mechanism of QcyP towards ALP. (b) The fluorescence emission spectra of QcyP with or without ALP. (c) The selectivity of QcyP (1: lysozyme; 2: PDE; 3: BSA; 4: avidin; 5: trypsin; 6: ACP; 7: esterase; 8: ALP). (d) Subcellular colocalization of QcyP. Reproduced with permission [52]. Copyright 2018, Elsevier. | |
Given ALP can remove phosphate groups from various substrates, this simple reaction offers infinite possibilities to design probes with kinds of luminescence mechanisms. It is believed that fluorophores with phosphatase reaction site for ALP detection continue to thrive. The organic probes for ALP detection which involved with kinds of chromophores are summarized in Table 1 [12, 21, 22, 33, 37, 42, 46, 47, 52-58]. These listed probes in Table 1 are all based on the dephosphorylation-induced fluorescence change. Due to the high molar extinction coefficients and easy modification, cyanines and its derivatives attracted huge scientific interest [51]. However, it should be noted that cyanine fluorophores suffer from poor stability to ROS, heat or light. More studies should be investigated on the modification of cyanine derivatives to improve their stability [59].
|
|
Table 1 Organic fluorescent probes for ALP detection based on the dephosphorylation-induced fluorescence change. |
FRET is a distance-dependent physical process. Through intermolecular long-range dipole-dipole coupling, energy is transferred from an excited molecular fluorophore (donor) to another fluorophore (receptor) without radiation [60]. The FRET- based ALP probe usually needs foreign substrates to react with the FRET pairs or linkers. Compared with dephosphorylation process, FRET probes gain more active sites to respond with ALP. And there are two main ways: ⅰ) The catalytic products could directly react with the donor or acceptor to cleave the FRET dyad and quench the fluorescence signal; ⅱ) The catalytic product could break the linker to separate the donor and acceptor, in these cases, the FRET is switched off.
Xiao et al. [18] utilized the fluorescent polydopamine (F-PDA) nanoparticles as the fluorescent donor and MnO2 nanosheets as the fluorescent acceptor to establish a label-free and low-cost ALP probe. Ascorbic acid 2-phosphate (AA-P) was chosen as the substrate of ALP. ALP could hydrolyze AA-P into AA, which could trigger the reduction and decomposition of MnO2 nanosheets into Mn2+, thereby it could terminate the FRET effect between F-PDA nanoparticles and MnO2 nanosheets, in this case, the fluorescence signal of F-PDA nanoparticles could be recovered. This FRET probe exhibited good sensing performance with a low detection limit of 0.34 mU/mL. In another case, Lu et al. [19] also established a FRET probe through the biocatalytic activity of ALP toward AA-P. In this work, the graphene quantum dots (GQDS) was used as the donor (Fig. 13a). GQDs has been discovered as a zero-dimensional graphitic nanocrystal with small size and tunable photoluminescence. The mechanism of this probe underlay in that AA could reduce the Ag+ into AgNPS, leading to the in-situ growth of AgNPS on the surface of GQDS. So when ALP was added, the AA-P could yield AA and further produce AgNPS on the surface of GQDS. Because of the FRET effect between attached AgNPS and GQDS, the fluorescence signal of GQDS was quenched at 461 nm which accompanied with a strong increase in the SPR band at 415 nm of AgNPS (Fig. 13b). As verified by transmission electron microscope (TEM) and atomic force microscopy (AFM) imagings, GQDS showed fairly uniform nanoparticles with the diameters about 3.3 nm. With the addition of ALP, AgNO3 and AA-P, the nanoparticles showed a larger diameter about 14.1 nm suggesting the formation of GQDS/AgNPS hybrid (Fig. 13c). This colorimetry and fluorescent dual-modality FRET probe exhibited high sensitivity of ALP with the detection limits of 0.02 U/L and 0.1 U/L by fluorometric and colorimetric measurements, respectively. As shown in Fig. 13d, in living cells imaging, strong fluorescence signal could be observed when incubation with GQDS and AA-P, but the fluorescence signal was disappeared with the addition of AgNO3. This dual-mode ALP detection assay provided a new insight in the visualization of ALP. In another case, Gorai et al. [61] established a pyrene-1-phosphate doped Eu-cholate gel to detect ALP. With the dephosphorylation, the energy transfer process could happen between pyrene and Eu3+, leading to strong fluorescence signal at 617 nm.

|
Download:
|
| Fig. 13. (a) Illustration of imaging mechanism based on FRET effect between AgNPS and GQDS triggered by ALP. (b) UV–vis absorption/fluorescence spectra of dual-mode sensing probe with different activities of ALP. (c) TEM images of GQDS (A) and GQDS/AgNPs hybrid (B), AFM images of GQDS (C) and GQDS/AgNPs hybrid (D). (d) Fluorescence imaging of probes in L0-2 cells. Reproduced with permission [19]. Copyright 2018, Elsevier. | |
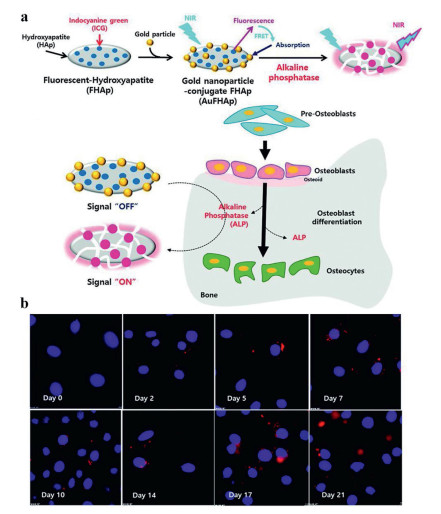
The efficiency of FRET is inversely proportional to the sixth power of the distance between donor and acceptor, which makes FRET very sensitive to even little changes in the distance between the donor and the acceptor [62]. In fact, the distance between this pair of donor and acceptor should be generally less than 10 nm. The changes in the distance between the donor and the acceptor can be used as the switch of FRET. Lim et al. [63] developed a nanoprobe by using hydroxyapatite (HAp) as the substrate to detect the level of ALP. Gold nanoparticle-conjugate fluorescent HAp (AuFHAp) was formulated with gold nanoparticles, indocyanine green (ICG) and HAp. HAp is a biocompatible material and carrier which could attach ICG and gold nanoparticles to its surface. Due to the absorption spectrum of gold nanoparticles was partially overlapped with the fluorescence of ICG, thus, gold nanoparticles as a acceptor could quench the fluorescence signal of ICG by FRET effect, leading to no fluorescence signal of AuFHAp. The design of FRET-based probes was based on the changes of the donor-acceptor distances induced by ALP. As shown in Fig. 14a, when ALP existed, the support and carrier role of HAp was destroyed, resulting in the separation of ICG and gold nanoparticles, leading to FRET failure. Thus, a remarkable fluorescence signal of ICG could be detected again. Considering the ALP activity usually gets stronger in accordance with the extent of osteogenic differentiation, the as-prepared AuFHAp was used to measure ALP activity in MC3T3-E1 cell during differentiation. As shown in Fig. 14b, the fluorescence signal of ICG gradually increased along with the culture time lasting, suggesting these cells had a good osteogenic differentiation. And these results were highly accordance with the data tested from cell lysates. In this case, AuFHAp provided a feasible tool to non-invasively monitor the ALP activity in bone tissues.

|
Download:
|
| Fig. 14. (a) Illustration of AuFHAp NPS for ALP detection. (b) Detection of ALP activity during osteoblast differentiation in MC3T3-E1 cells. Blue filter for cell nuclei (Hoechst 33, 324) and a red filter for ICG. Reproduced with permission [63]. Copyright 2015, Royal Society of Chemistry. | |
Internal filtering effect (IFE) is a unique phenomenon in fluorescence spectroscopy. When coexist with other substances (absorber) whose absorption spectra overlaps with excitation or emission spectras of fluorophores, the fluorescence signal of fluorophores gets week or even quenched. The striking advantage of IFE probe is no surface modification or covalent linking for fluorophores, giving cost-effective and simple probe fabrication [64]. Compared with ICT and PeT probes, no intramolecular interaction was needed for IFE probes, making the IFE-based detection more flexible [64]. The IFE effect has become an effective strategy for the design and development of fluorescent probes. However, the overlap between the spectrums of absorber and fluorophore is not easy to achieve, leading to limited application of IFE probes. In this case, quantum dots (QDS), whose emission peaks are easy to adjust by changing the size, became the most suitable fluorescence probes based on IFE. Recently, several on-off/off-on IFE probes based on QDS have been developed for ALP detection [17].
QDS are nano-level semiconductors. Compared with organic fluorescent dyes, QDs have better photostability and narrow half-peak width, which is more conducive to multi-channel detection or imaging applications. Due to the quantum confinement effect, the continuous energy band of QDS splits into separate energy levels, which can generate fluorescence emission under the excitation of light [65]. Importantly, QDS possess different emission peaks from the visible light region to the near-infrared light region. QDS-based ALP probes commonly utilized PNPP or its enzyme catalytic product p-nitrophenol (PNP) as the candidate absorber [17]. The IFE-based ALP probes with PNPP as substrate are summarized in Table 2 [16, 66-76]. When incubation with ALP, PNPP could be degraded into PNP, leading to the red-shift of absorption band from 310 nm to 404 nm. The basic point is that the absorption of PNPP or PNP should overlap with excitation spectras of QDS, and thus induce the fluorescence signal emission or quenching.
|
|
Table 2 Fluorescent probes for ALP detection based on the PNPP (absorber). |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Recently, Li et al. [66] designed N-doped carbon dots (CDs) with quantum yield of 49%. And PNPP was utilized to be the substrate of ALP. Due to the competitive absorption between CDs and PNP, the excitation of CDs was enormously weakened with the gradual degradation of PNPP under the catalysis of ALP. In this case, a turn-off phosphorescence assay was developed for ALP detection. Considering the abnormal level of ALP was related to many deseases, CDS were tested in human serum samples. This probe could accurately detect ALP level in serum with the detection limit of 0.01 U/L. Importantly, CDs were also successfully applied to ALP cell imaging. The fluorescence intensity from RAW 264.7 cells decreased gradually with the prolonging of time due to the IFE effect between CDs and PNP. These results were impressive which implyed that the IFE-based ALP probes provide a potential platform for ALP sensing in living cell imaging and clinical diagnosis.
3. Conclusions and outlookIn the past few years, phosphatase probes have been widely explored for disease diagnosis. In this review, we made a clear classification according to their luminescence mechanisms, including ICT, AIE, PeT, ESIPT, rearrangement of conjugation system, FRET and IFN. Taking advantages of fluorescence imaging, these ALP probes could detect the analyte in a real-time way. However, most of these detections are limited to cellular level and human blood samples in vitro [64, 77]. The sensitivity and selectivity of ALP probes are rarely evaluated in vivo. Therefore, it is highly desirable to estabilsh ALP-related animal models for in vivo evaluation of ALP probes. Meanwhile, the systematic assessment of their toxicity, biocompatibility, immunogenicity, pharmacokinetics, and biodistribution will be critical.
The abnormal levels of ALP are related to many diseases, so quantitative detection of ALP level is crucial [6]. However, because of the same catalytic mechanism of ALP and ACP, the ALP probe usually could respond to ACP at suitable pH. Although the content of ACP is much less than ALP in human, but it still influences the accurate quantification of ALP. Although some probes could distinguish ALP from ACP at specific pH, most of these selectivity tests are performed at PBS with the pH at 7.4 for mimicking the physiological environment. Considering the pH is very important for the biocatalytic activity of ALP and ACP, and the pH of lysosomes is usually below 5.5, thus more wide range of pH needs to be tested. In addition, electrophoresis which is time-consuming and laborious is the main method to distinguish the isoenzyme of ALP. But how to distinguish the isoenzymes of ALP via fluorescence imaging remains poorly understand. Thus, the selectivity of ALP fluorescent probes needs to be improved in the future.
As mentioned above, off-on, on-off and ratiometric strategies are involved in the detection of ALP, and among them, off-on strategy is the most frequently used method. However, the off-on strategy is easily affected by many factors, thus leading to inaccurate quantification of ALP. In comparison, ratiometric probes could largely increase the reliability and accuracy of detection. Thus, more ratiometric ALP probes with high sensitivity and selectivity need to be designed and developed in the future [37].
Both organic and inorganic materials have their weaknesses, such as short fluorescence lifetimes and limited solubility for organic ALP probes and poor biocompatibility for inorganic probes. Besides, the luminescence mechanism is lmited to the dephosphorylation reaction and corporatation with foreign labels. However, as mentioned before, phosphate ester is usually substrates of other phosphatases in addition to ALP, thus, the development of promising materials and novel luminescence mechanisms will be of great help for ALP specific detection.
In summary, many fluorescent probes have been developed for ALP detection. ALP fluorescent probes have made a good progress and achievement in cell imaging and real-time monitoring. But in the future, many issues still need to concern for ALP fluorescent probes in the clinical application. There is no doubt that ALP fluorescent probes will play significant roles in ALP-related disease diagnosis.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is financially supported by National Key Research and Development Program (No. 2018YFA0704003), Basic Research Program of Shenzhen (Nos. JCYJ20170412111100742, JCYJ20180507182413022), Fok Ying-Tong Education Foundation for Young Teachers in the Higher Education Institutions of China (No. 161032), and Guangdong Province Natural Science Foundation of Major Basic Research and Cultivation Project (No. 2018B030308003).
| [1] |
P. Ou, R. Zhang, Z. Liu, et al., Angew. Chem. Int. Ed. 58 (2019) 2261-2265. DOI:10.1002/anie.201811391 |
| [2] |
L. Li, J. Ge, H. Wu, et al., J. Am. Chem. Soc. 134 (2012) 12157-12167. DOI:10.1021/ja3036256 |
| [3] |
A. Krzyzosiak, A. Sigurdardottir, L. Luh, et al., Cell 174 (2018) 1216-1228. DOI:10.1016/j.cell.2018.06.030 |
| [4] |
J. Brognard, E. Sierecki, T. Gao, A.C. Newton, Mol. Cell 25 (2007) 917-931. DOI:10.1016/j.molcel.2007.02.017 |
| [5] |
L. Dong, J. Qian, Z. Hai, et al., Anal. Chem. 89 (2017) 6922-6925. DOI:10.1021/acs.analchem.7b00621 |
| [6] |
Y. Han, J. Chen, Z. Li, H. Chen, H. Qiu, Biosens. Bioelectron. 148 (2020) 111811. DOI:10.1016/j.bios.2019.111811 |
| [7] |
S.M. Wasif, D. Alexander, C.M. Wicox, J. Appl. Res. 5 (2005) 88-95. |
| [8] |
T.P. Gade, M.W. Motley, B.J. Beattie, et al., PLoS One 6 (2011) e22608. DOI:10.1371/journal.pone.0022608 |
| [9] |
A.L. Brichacek, C.M. Brown, Metab. Brain Dis. 34 (2019) 3-19. DOI:10.1007/s11011-018-0322-3 |
| [10] |
H. Jiao, J. Chen, W. Li, et al., ACS Appl. Mater. Interfaces 6 (2014) 1979-1985. DOI:10.1021/am405020b |
| [11] |
R. Kong, T. Fu, N. Sun, et al., Biosens. Bioelectron. 50 (2013) 351-355. DOI:10.1016/j.bios.2013.06.064 |
| [12] |
S. Li, C. Li, Y. Li, et al., Anal. Chem. 89 (2017) 6854-6860. DOI:10.1021/acs.analchem.7b01351 |
| [13] |
F.R. Baptista, S.A. Belhout, S. Giordani, S.J. Quinn, Chem. Soc. Rev. 44 (2015) 4433-4453. DOI:10.1039/C4CS00379A |
| [14] |
S.M. Albillos, R. Reddy, R. Salter, J. Food Protect. 74 (2011) 1144-1154. DOI:10.4315/0362-028X.JFP-10-422 |
| [15] |
H.N. Fernley, S. Bisaz, Biochem. J. 107 (1968) 279-283. DOI:10.1042/bj1070279 |
| [16] |
G. Zheng, X. Zhu, S. Wang, et al., Nanoscale 10 (2018) 19579-19585. DOI:10.1039/C8NR05767B |
| [17] |
R. Freeman, T. Finder, R. Gill, I. Willner, Nano Lett. 10 (2010) 2192-2196. DOI:10.1021/nl101052f |
| [18] |
T. Xiao, J. Sun, J. Zhao, et al., ACS Appl. Mater. Interfaces 10 (2018) 6560-6569. DOI:10.1021/acsami.7b18816 |
| [19] |
H. Lu, M. Zhang, D. Wu, et al., Sens. Actuators B: Chem. 258 (2018) 461-469. DOI:10.1016/j.snb.2017.11.122 |
| [20] |
X. Niu, K. Ye, L. Wang, et al., Anal. Chem. 1086 (2019) 29-45. |
| [21] |
L. Gwynne, A.C. Sedgwick, J.E. Gardiner, et al., Front. Chem. 7 (2019) 255. DOI:10.3389/fchem.2019.00255 |
| [22] |
Y. Tan, L. Zhang, K.H. Man, et al., ACS Appl. Mater. Interfaces 9 (2017) 6796-6803. DOI:10.1021/acsami.6b14176 |
| [23] |
Q. Zhang, S. Li, C. Fu, et al., J. Mater. Chem. B: Mater. Biol. Med. 7 (2019) 443-450. DOI:10.1039/C8TB02799D |
| [24] |
R.L. Dean, Biochem. Mol. Biol. Educ. 30 (2002) 401-407. DOI:10.1002/bmb.2002.494030060138 |
| [25] |
J. Wang, J. Zhou, H. He, et al., Chembiochem 20 (2019) 526-531. DOI:10.1002/cbic.201800495 |
| [26] |
L. Li, X. Shen, Q. Xu, S.Q. Yao, Angew. Chem. Int. Ed. 52 (2013) 424-428. DOI:10.1002/anie.201205940 |
| [27] |
X. Gao, G. Ma, C. Jiang, et al., Anal. Chem. 91 (2019) 7112-7117. DOI:10.1021/acs.analchem.9b00109 |
| [28] |
D. Yao, S. Yang, Y. Wang, et al., Nanoscale 11 (2019) 6307-6314. DOI:10.1039/C9NR00913B |
| [29] |
Y. Gao, J. Shi, D. Yuan, B. Xu, Nat. Commun. 3 (2012) 1033-1040. DOI:10.1038/ncomms2040 |
| [30] |
L. Dong, Q. Miao, Z. Hai, et al., Anal. Chem. 87 (2015) 6475-6478. DOI:10.1021/acs.analchem.5b01657 |
| [31] |
J. Zhan, Y. Cai, S. He, et al., Angew. Chem. Int. Ed. 57 (2018) 1813-1816. DOI:10.1002/anie.201710237 |
| [32] |
R. Yan, Y. Hu, F. Liu, et al., J. Am. Chem. Soc. 141 (2019) 10331-10341. DOI:10.1021/jacs.9b03649 |
| [33] |
X. Gu, G. Zhang, Z. Wang, et al., Analyst 138 (2013) 2427-2431. DOI:10.1039/c3an36784c |
| [34] |
Y. Cui, R. Zhang, L. Yang, S. Lv, Chin. Chem. Lett. 30 (2019) 1078-1082. DOI:10.1016/j.cclet.2018.10.017 |
| [35] |
H. Li, Q. Yao, F. Xu, et al., Angew. Chem. Int. Ed. 59 (2020) 2-12. DOI:10.1002/anie.201914768 |
| [36] |
J. Deng, P. Yu, Y. Wang, L. Mao, Anal. Chem. 87 (2015) 3080-3086. DOI:10.1021/ac504773n |
| [37] |
Z. Song, R.T.K. Kwok, E. Zhao, et al., ACS Appl. Mater. Interfaces 6 (2014) 17245-17254. DOI:10.1021/am505150d |
| [38] |
C. Fan, S. Luo, H. Qi, Luminescence 31 (2016) 423-427. DOI:10.1002/bio.2977 |
| [39] |
T. Teshima, M. Takeishi, T. Arai, New J. Chem. 33 (2009) 1393-1401. DOI:10.1039/b823431k |
| [40] |
A.C. Sedgwick, L. Wu, H. Han, et al., Chem. Soc. Rev. 47 (2018) 8842-8880. DOI:10.1039/C8CS00185E |
| [41] |
T.I. Kim, H. Kang, G. Han, et al., Chem. Commun. (Camb.) (2009) 5895-5897. |
| [42] |
J. Liang, R.T.K. Kwok, H. Shi, et al., ACS Appl. Mater. Interfaces 5 (2013) 8784-8789. DOI:10.1021/am4026517 |
| [43] |
P. Zhang, F. Sun, C. Tsao, et al., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 12046-12051. DOI:10.1073/pnas.1512465112 |
| [44] |
J.F. Callan, A.P. de Silva, D.C. Magri, Tetrahedron 61 (2005) 8551-8588. DOI:10.1016/j.tet.2005.05.043 |
| [45] |
T. Terai, K. Kikuchi, S.Y. Iwasawa, et al., J. Am. Chem. Soc. 128 (2006) 6938-6946. DOI:10.1021/ja060729t |
| [46] |
H. Zhang, C. Xu, J. Liu, et al., Chem. Commun. (Camb.) 51 (2015) 7031-7034. DOI:10.1039/C5CC01005E |
| [47] |
H. Zhang, P. Xiao, Y. Wong, et al., Biomaterials 140 (2017) 220-229. DOI:10.1016/j.biomaterials.2017.06.032 |
| [48] |
T.I. Kim, M.S. Jeong, S.J. Chung, Y. Kim, Chemistry 16 (2010) 5297-5300. DOI:10.1002/chem.201000154 |
| [49] |
W. Shi, H. Ma, Chem. Commun. (Camb.) 48 (2012) 8732-8744. DOI:10.1039/c2cc33366j |
| [50] |
A. Podder, S. Senapati, P. Maiti, et al., J. Mater. Chem. B: Mater. Biol. Med. 6 (2018) 4514-4521. DOI:10.1039/C8TB01143E |
| [51] |
C. Sissa, A. Painelli, F. Terenziani, et al., Phys. Chem. Chem. Phys. 22 (2019) 129-135. |
| [52] |
Z. Gao, J. Sun, M. Gao, et al., Sens. Actuators B: Chem. 265 (2018) 565-574. DOI:10.1016/j.snb.2018.03.078 |
| [53] |
H. Liu, K. Li, X. Hu, et al., Angew. Chem. Int. Ed. 56 (2017) 11788-11792. DOI:10.1002/anie.201705747 |
| [54] |
X. Zhou, Y. Jiang, X. Zhao, Y. Zhu, Molecules 21 (2016) 1619-1628. DOI:10.3390/molecules21121619 |
| [55] |
P. Zhang, C. Fu, Q. Zhang, et al., Anal. Chem. 91 (2019) 12377-12383. DOI:10.1021/acs.analchem.9b02917 |
| [56] |
T.I. Kim, H. Kim, Y. Choi, Y. Kim, Chem. Commun. (Camb.) 47 (2011) 9825-9827. DOI:10.1039/c1cc13819g |
| [57] |
Z. Lu, J. Wu, W. Liu, et al., RSC Adv. 6 (2016) 32046-32051. DOI:10.1039/C6RA00983B |
| [58] |
H. Liu, X. Hu, L. Zhu, et al., Talanta 175 (2017) 421-426. DOI:10.1016/j.talanta.2017.04.081 |
| [59] |
N. Karton-Lifshin, E. Segal, L. Omer, et al., J. Am. Chem. Soc. 133 (2011) 10960-10965. DOI:10.1021/ja203145v |
| [60] |
A. Xu, L. Zhang, H. Zeng, et al., Mikrochim. Acta 185 (2018) 288-295. DOI:10.1007/s00604-018-2827-1 |
| [61] |
T. Gorai, U. Maitra, J. Mater. Chem. B: Mater. Biol. Med. 6 (2018) 2143-2150. DOI:10.1039/C7TB02657A |
| [62] |
X. Zhang, Y. Hu, X. Yang, et al., Biosens. Bioelectron. 138 (2019) 111314-111326. DOI:10.1016/j.bios.2019.05.019 |
| [63] |
E.K. Lim, J.O. Keem, H.S. Yun, et al., Chem. Commun. (Camb.) 51 (2015) 3270-3272. DOI:10.1039/C4CC09620G |
| [64] |
G. Mao, Q. Zhang, Y. Yang, et al., Anal. Chem. 1047 (2019) 208-213. |
| [65] |
S. Takano, S. Hasegawa, M. Suyama, T. Tsukuda, Acc. Chem. Res. 51 (2018) 3074-3083. DOI:10.1021/acs.accounts.8b00399 |
| [66] |
J. Zhang, X. Lu, Y. Lei, et al., Nanoscale 9 (2017) 15606-15611. DOI:10.1039/C7NR03673F |
| [67] |
G. Li, H. Fu, X. Chen, et al., Anal. Chem. 88 (2016) 2720-2726. DOI:10.1021/acs.analchem.5b04193 |
| [68] |
M. Mao, T. Tian, Y. He, et al., Mikrochim. Acta 185 (2017) 17-23. |
| [69] |
C. Tang, Z. Qian, Y. Huang, et al., Biosens. Bioelectron. 83 (2016) 274-280. DOI:10.1016/j.bios.2016.04.047 |
| [70] |
Q. Liu, H. Li, R. Jin, et al., Sens. Actuators B: Chem. 281 (2019) 175-181. DOI:10.1016/j.snb.2018.10.083 |
| [71] |
X. Ren, Z. Chen, X. Chen, et al., J. Lumin. 145 (2014) 330-334. DOI:10.1016/j.jlumin.2013.07.069 |
| [72] |
Y. Zhong, F. Xue, P. Wei, et al., Nanoscale 10 (2018) 21298-21306. DOI:10.1039/C8NR05549A |
| [73] |
L. Jia, J.P. Xu, D. Li, et al., Chem. Commun. (Camb.) 46 (2010) 7166-7168. DOI:10.1039/c0cc01244k |
| [74] |
A.K. Sharma, S. Pandey, M.S. Khan, H.F. Wu, Sens. Actuators B: Chem. 259 (2018) 83-89. DOI:10.1016/j.snb.2017.11.190 |
| [75] |
H. Liu, M. Li, Y. Xia, X. Ren, ACS Appl. Mater. Interfaces 9 (2017) 120-126. DOI:10.1021/acsami.6b11920 |
| [76] |
N. Malashikhina, G. Garai-Ibabe, V. Pavlov, Anal. Chem. 85 (2013) 6866-6870. DOI:10.1021/ac4011342 |
| [77] |
L. Xu, X. He, Y. Huang, et al., J. Mater. Chem. B: Mater. Biol. Med. 7 (2019) 1284-1291. DOI:10.1039/C8TB03230K |