 2021, Vol. 32
2021, Vol. 32 


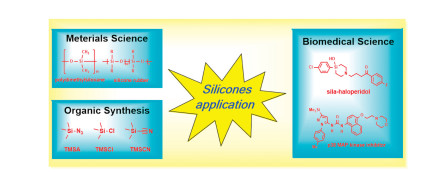
As we all know, silicon is the second largest element (26.4%) in the earth's crust. Such a large reserve also facilitates human to exploring and utilizing silicon compounds. Organosilicon compounds have been greatly developed in the past 100 years. With their advantages of low toxicity, fireproof insulation, low cost and easy availability, high and low temperature resistance, anti-aging, hydrophobic and flame retardant [1], they have been widely used in various fields, such as material science [2], medicine [3] and organic synthesis [4-6] (Fig. 1). Silicon materials are widely used in all aspects of human life, such as electronic appliances, environmental energy, personal care, biomedical, aerospace and construction industries [7]. In organic synthesis, silicon reagents have a number of applications, such as protective reagents for carbonyl and hydroxyl groups, electrophilic reagents [8], precursors of silicon radicals [9]. Furthermore, some scientists have discovered that silicon element also plays an important role in the process of human bone development and metabolism [10, 11].

|
Download:
|
| Fig. 1. Applications of organosilicons. | |
{kind=link}
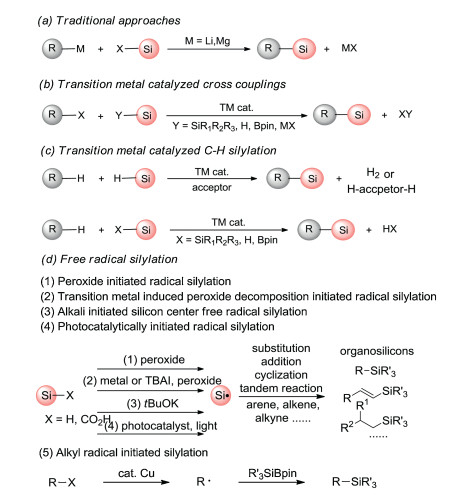
Traditionally, the protocols for preparing organosilicon derivatives are achieved through the nucleophilic reactions of organometallic reagents (grignard reagents, organolithium reagents, organozinc reagents) with chlorosilanes or epoxy silanes (Scheme 1a), but these methods suffer from some limitations including poor group tolerance, the use of toxic and unstable reagents, harsh conditions and poor atom economy [12, 13]. Later, researchers develop transition metal-catalyzed synthesis of organosilicon compounds through the cross-coupling of halogenated hydrocarbons or hydrocarbons with silylation reagents [14-18]. The former (Scheme 1b) requires pre-activation of substrates. The latter (Scheme 1c) has a great advantage in atomic economy owing to the directly C-H silylation, but the reaction selectivity is unsatisfactory. Meanwhile, harsh conditions, the use of stoichiometric additives and expensive transition-metal catalysts are norm in these protocols.

|
Download:
|
| Scheme 1. The measures for the synthesis of organic silicon compounds. | |
{kind=link}
As we all know, free radicals has highly chemically reactivity and unique stability, so the preparation of organic compounds by free radical reaction usually has the advantages of high yield, relatively mild conditions, good functional group tolerance and selectivity [19-22]. Thus, the synthesis of organosilicons by the free radical routes has also received much attention in recent years [23-26]. In the view of these investigations, we summarize five main free-radical initiation pathways for the formation of organosilicons including (1) metal-free peroxide initiated radical silylation; (2) transition-metal-induced peroxide decomposition initiated radical silylation; (3) alkali initiated radical silylation; (4) photocatalytically initiated radical silylation; (5) alkyl radical initiated silylation (Scheme 1d). Although silyl radicals triggered by peroxide, electron-induced peroxide decomposition, alkali, photocatalysis, can achieve a number of radical silylations, they fail to activate the C(sp3)-X (X is any element) bonds. An alternative for the generation of C(sp3)-Si bonds is disclosed, in which alkyl radicals can initiate radical silylation.
In the view of the importance of organosilicons and the advantages of radical silylations, several excellent reviews on this area have been published. In 2016, Shang and coworkers [23] briefly review the research on free radical-promoted selective C-H/Si-H functionalization to form C-Si bond. They divide radical silylation reactions according to the initiation ways of silyl free radical: (1) peroxide promotes free radical addition-dehydrogenation process and (2) alkali promotes free radical addition-dehydrogenation process. In 2018, Li and coworkers [24] first reviewed the research progress of photo-mediated generation of silicon-based free radicals, and their applications in organic synthesis. In 2019, two reviews on silylation reactions addressed the transition-metal-catalyzed radical silylation of C-H bonds [25] and copper-catalyzed radical silylation [26].
According to above reviews, there is no systematic overview of the research progress on the synthesis of organosilicon by the free radical pathway. Therefore, this review will supplement the content not covered by the above reviews, mainly introducing the new free radical methods for the synthesis of organosilicons since 2016. Our aim is to stimulate further research on new free radical routes to organosilicons and new catalytic systems for these transformations.
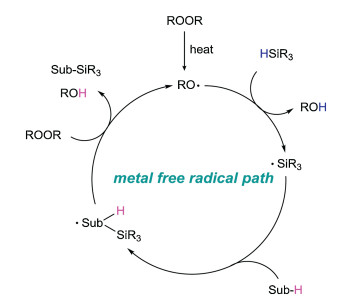
2. Peroxide initiated radical silylationA tentative mechanism of this radical silylation reaction is illustrated in Fig. 2. Firstly, peroxide is decomposed into two alkoxy radicals (or a molecule of alkoxy free radical and a molecule of hydroxyl radical) under heating conditions. Then, silicon center free radical is generated by hydrogen atom transfer (HAT) bewteen alkoxy radical and hydrogen silane, following the silylaton of substrate. Finally, the product are yielded by HAT process. In addition, the generated alkoxy radical enters the next cycle to continue the radical reaction.

|
Download:
|
| Fig. 2. Peroxide initiated radical silylation mechanism. | |
{kind=link}
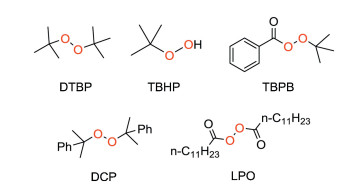
The use of different peroxides here has a great influence on the reaction, because they determine the rate of silicon radicals generation which is the key step for the silylation [23]. The commonly used peroxide initiators are DTBP, TBHP, TBPB, DCP, LPO (Fig. 3) and inorganic peroxides (Na2S2O8, K2S2O8).

|
Download:
|
| Fig. 3. Commonly used peroxides. | |
{kind=link}
Li and co-workers [27] reported on the synthesis of silafluorene (Scheme 2a) and silaindene compounds (Scheme 2b) using arylhydrosilane as the raw material via the intramolecular or intermolecular cyclization under metal-free and neutral conditions. This method furnishes a wide range of products with high yields via direct Si-H and C-H cleavage pathways in one step, thus representing a simple and effective way for the synthesis of benzosilane derivatives.

|
Download:
|
| Scheme 2. Synthesis of silicon based fluorenes and indenes compounds by intramolecular and intermolecular cyclization. | |
{kind=link}
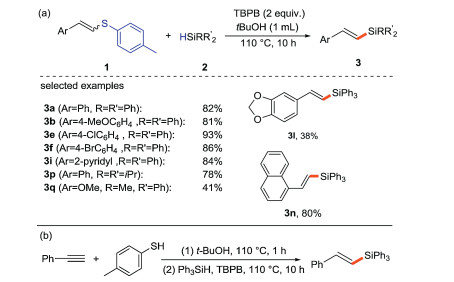
Our group [28] developed a free radical-mediated method for the activation and cleavage of C(sp2)-S bonds to synthesize alkenyl silanes. These free radical substitutions are carried out under metal-free conditions and only E-products are observed in all cases. The authors firstly activate the C(sp2)-S bonds through radical pathway for the construction of C(sp2)-Si bonds (Scheme 3a). A series of arylalkenyl sulfides with electron-donating and electron-withdrawing groups provide medium to excellent yields (3a, 3b, 3e, 3f, 3l), and the reactions of heteroaryl and naphthylalkenyl sulfides are also carried out smoothly (3i, 3n). Moreover, different silanes (3p, 3q) can be applied to this protocol. This method also realizes the stereospecific silylation reaction of acetylene benzene through a one-pot two-step route to obtain E-isomer 3a (Scheme 3b).

|
Download:
|
| Scheme 3. Synthesis of alkenyl silane by CS bond activation of aryl alkenyl sulfide. | |
{kind=link}
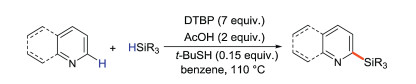
Maruoka's group [29] reported a free radical reaction of pyridine (or other N-heterocycles) with hydrosilane under transition-metal-free conditions, which accomplished the direct C-H bond silylation of electron-deficient N-heteroaromatics in the presence of acetic acid, DTBP and tert-butylthiol (Scheme 4). This approach allows the direct and selective introduction of trialkylsilyl at the ortho-position of pyridine rings.

|
Download:
|
| Scheme 4. Silylation of N-heterocycles. | |
{kind=link}
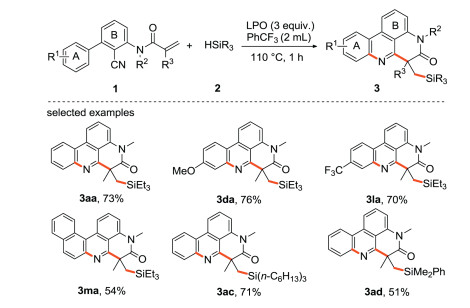
Liu and Sun et al. [30] reported a metal-free, cascade transformation for the synthesis of silyl functionalized 4H-pyrido[4, 3, 2-g'h]phenanthridin-5(6H)-one using several readily available hydrosilanes as silicon sources (Scheme 5). Both substrates containing the electron-donating and electron-withdrawing groups on the benzene ring A provide the desired products (3aa, 3da, 3la) in moderate yields (55%–76%). The reaction can also take place smoothly (3ma) in the case of naphthyl substituted reactant. As expected, both isopropyl silane and trihexyl silane can react with 1 to get the desired products (3ac, 3ad).

|
Download:
|
| Scheme 5. The cascade cyclization for the synthesis of silyl functionalized 4H-pyrido [4, 3, 2-gh]phenanthridin-5(6H)-ones. | |
{kind=link}
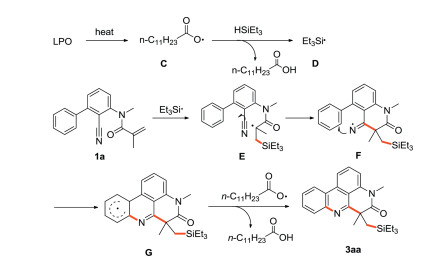
A possible free radical addition/cyano insertion/homolytic aromatic substitution (HAS) cascade mechanism is proposed (Scheme 6). First, the thermal decomposition of LPO generates free radical C, and then C extracts a hydrogen radical from triethylsilane to generate silyl radical D. The selective addition of D with carbon-carbon double bond generates radical intermediate E, following intramolecular cyclization with cyano group to obtain imino radical F which is conjugated to the benzene ring via HAS. Finally, C extracts hydrogen radicals from intermediate G to provide the desired product 3aa.

|
Download:
|
| Scheme 6. Silyl radical initiated cascade addition/cyclization mechanism. | |
{kind=link}
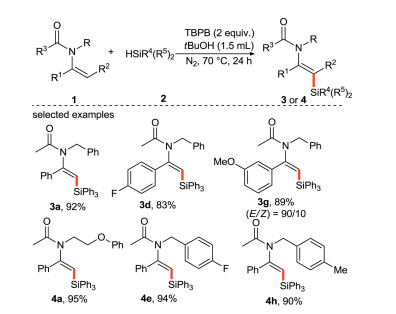
In 2020, Xu's group [31] developed a new method for metal-free dehydrogenative silylation of enamides, affording the desired functionalization alkenyl silane products with high yields and stereoselectivity (Scheme 7). This protocol also displays good tolerance of various functionalities. Notably, these products can be transformed to other useful compounds, demonstrating the highly synthetic utility of this methodology.

|
Download:
|
| Scheme 7. Metal-free dehydrosilylation of enamides. | |
{kind=link}
3. Transition metal induced peroxide decomposition initiated radical silylation
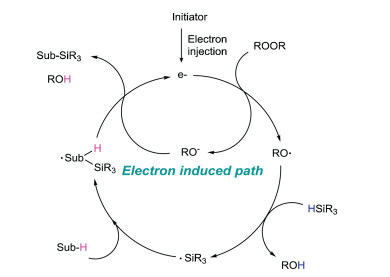
Different from the pathway of radical silylation initiated by peroxides, this synthetic method usually requires transition metal catalysts as electron donors to initiate free radical reactions, so the selection of catalysts (electron-donors) is very important. At present, the main electron-donors used in the reaction are copper compounds, such as Cu2O, CuCl, Cu(OAc)2, CuO owing to their excellent electron transfer capability [26]. Iron compounds, TBAI and photocatalysts can also be used as the electron-donors to optimize the reaction conditions.
A tentative mechanism of this radical silylation is illustrated in Fig. 4. Electron donor provides an electron to initiate the cycle system. After accepting the electron, the peroxide cleaves into a molecular alkoxy radical and a molecular alkoxy anion. The alkoxy radical extracts hydrogen from hydrosilane to generate silicon center free radical. This silyl radical is added to the substrate to form a free radical intermediate, which reacts with the previous peroxide to produce alcohols and silylation products, as well as produce an electron feed cycle system.

|
Download:
|
| Fig. 4. The mechanism of electron-induced peroxide decomposition initiated radical silylation. | |
{kind=link}
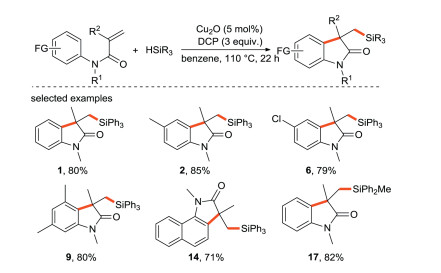
The research group of Liu [32] reported an approach for the selective activation of Si-H/C-H bonds to achieve the silylation reaction (Scheme 8), which offered an efficient access to silylated oxindoles through a free-radical cascade process. In this route, Cu2O is applied as the electron donor, and DCP is used as the free radical initiator.

|
Download:
|
| Scheme 8. Silylation of activated olefin and silane. | |
{kind=link}
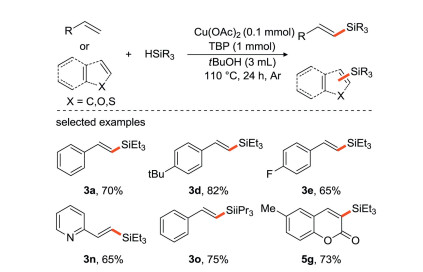
In 2016, our group [33] developed a copper-catalyzed direct silylation of olefins and silanes to synthesize alkenyl organosilicon compounds with highly stereoselectivity (Scheme 9), which provided a new and convenient strategy for the synthesis of useful alkenyl silanes. Moreover, the protocol is proposed to proceed via a radical pathway and exhibits good functional group tolerance.

|
Download:
|
| Scheme 9. Copper-catalyzed silylation of olefins and silanes to synthesize alkenyl silanes. | |
{kind=link}
The Liu's group [34] reported a stereospecific silylation reaction of acrylic acid or propanoic acid with silane via radical addition/decaroxylation processes. This is the first example of decarboxylation for the construction of C-Si bonds (Scheme 10), providing an effective and simple way for the preparation of various alkenyl and alkynyl organosilicon compounds from α, β-unsaturated acids. The decarboxylation has also attracted much attention due to the use of carboxylic acids as substrates instead of halides and organometallic reagents.

|
Download:
|
| Scheme 10. Decarboxylation silylation of acrylic acid and propanoic acid with silane. | |
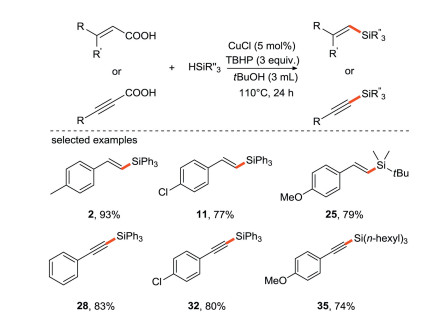{kind=link}
Loh and Xu et al. [35] developed a copper-catalyzed alkenes with α, β-unsaturated compounds or conjugated enynes to form bifunctional silicon-based peroxidation reaction (Scheme 11). Various silicon-based peroxidation products were directly obtained in moderate to good yields with good tolerance of functional groups. This strategy provides an effective way for the preparation of multi-functional silicon peroxides by one-step method.

|
Download:
|
| Scheme 11. Silicon-based peroxidation of CC bond with α, β-unsaturated ketones, esters, amides and conjugated alkynes. | |
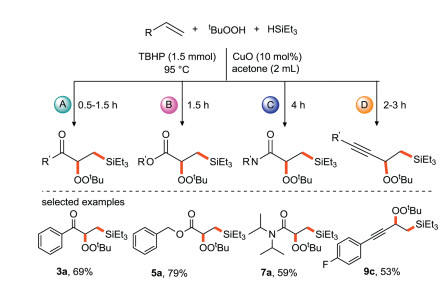{kind=link}
The Zhu research group [36] used dicumyl peroxide (DCP) as an initiator to comply the Cu-catalyzed radical silylarylation of acetylenic ketone with silane (Scheme 12) for the preparation of silicon-based functionalized indenones, which mainly relied on Rh-catalyzed carbonylation/cyclization of silicon alkyne and specific arylboronic acid in the past. This chemistry has the characteristics of good selectivity, the ability of gram-scale magnification, broad substrate scope and easy availability of starting materials, thus providing an effective way for the synthesis of silicon-based functionalized indenone derivatives.

|
Download:
|
| Scheme 12. Free radical silylarylation of alkynone. | |
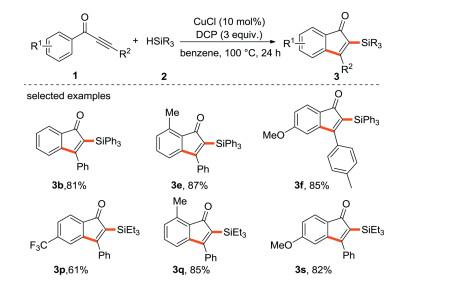{kind=link}
Luo and Li et al. [37] reported the intermolecular 1, 2-difunctionalization of styrenes or conjugated alkenes with silanes and nucleophiles for the first time. Classically, the synthesis of 1-amino-2-silylalkanes and other silicon-containing amino compounds depends on the introduction of either the amino or silicon functional groups into the corresponding silicon-based or amino-based frameworks. These methods have harsh reaction conditions such as the use of strong bases, narrow substrates scope, multi-step synthetic route or the use of expensive silicon reagents. Herein, the authors developed an iron/peroxide initiated radical process to compound 1-amino-2-silylalkane bifunctional products (Scheme 13a). Moreover, the approach is also extended to the 1, 2-bifunctional silylation of olefins with other nucleophiles including indoles, pyrroles and 1, 3-dicarbonyls (Scheme 13b). This method has good functional group tolerance, so it is a powerful platform for the production of various multifunctional silanes.

|
Download:
|
| Scheme 13. Iron-catalyzed intermolecular 1, 2-difunctionalization of alkenes with silanes and nucleophiles. | |
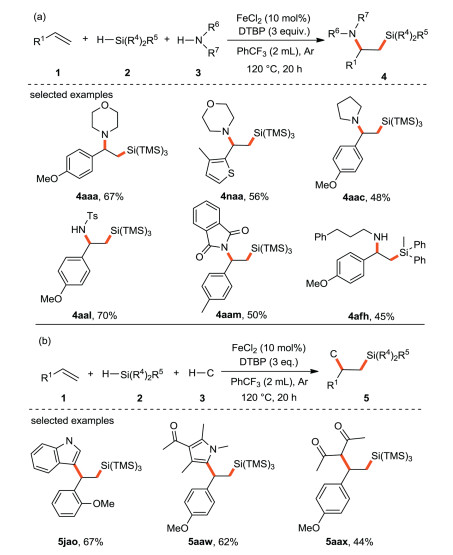{kind=link}
A possible mechanism of this approach is proposed (Scheme 14). First, DTBP is heated and cleaved by active FeII to provide alkoxy radical tBuO· and FeIII (tBuO). Silicon-center radical A is obtained by hydrogen atom transfer of tert-butyl oxygen radical and 2a, then alkyl radical intermediate B is obtained by addition of A with olefin 1a. FeIII(tBuO) oxidizes B to produce alkyl cations C and tert-butyl oxygen anions, and FeII is obtained to enter the next catalytic cycle. Nucleophiles and tBuO- undergo hydrogen atom transfer, and bond with C to give product 4 or 5.

|
Download:
|
| Scheme 14. Reaction mechanism of 1, 2-difunctionalization of alkenes with silanes and nucleophiles. | |
{kind=link}
Liu's group [38] reported the Cu/DTBP-promoted C-H radical silylation of arenes in 2017. The results suggest that both electron-rich and electron-poor aromatics can provide the final arylsilanes with good para-selectivity (Scheme 15a), which makes this free radical silylation to be an attractive strategy for the formation of C(sp2)-Si bonds. In addition, various heterocyclic compounds such as benzofuran, benzothiophene, thiophene, indole, pyridine and pyrrole, can be also applied in similar system expect that CuF2 in place of Cu2O is used as the catalyst (Scheme 15b).

|
Download:
|
| Scheme 15. Aryl/heteroaryl CH silylation reaction. | |
{kind=link}
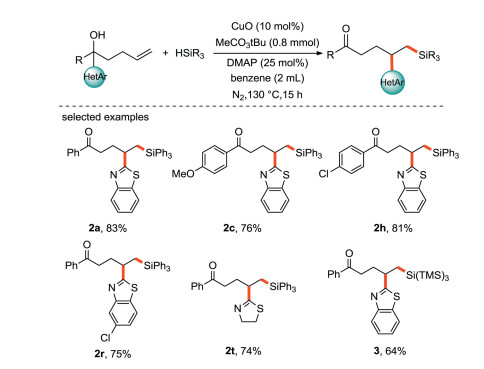
Zhu's group [39] reported a copper-catalyzed heteroarylsilylation of unactivated olefins via intramolecular remote migration of heteroaryl groups initiated by the addition of silyl free radical to C=C bonds (Scheme 16). Heteroaryl groups such as benzothiazolyl, thiazolyl, imidazolyl and pyridyl show their mobility, and various heteroaryl-substituted alkylsilanes are easily produced in good yields.

|
Download:
|
| Scheme 16. Heteroaryl silylation of unactivated olefins via internal and remote migration. | |
{kind=link}
In order to clarify the migration mechanism, the authors used a series of benzothiazole substituted tertiary alcohols with different alkyl chain lengths to conduct control experiments under standard conditions. Only bishomoallylic and trishomoallyl alcohols yield the corresponding migrated products (Scheme 17a). These results indicate that this kind of migration may be carried out in an intramolecular way, in which the five-membered or six-membered ring transition state is formed in the reaction of bishomoallylic or trishomoallyl alcohol. Based on the above experimental results, a possible mechanism is proposed (Scheme 17b). First of all, tert-butyl peracetate decomposes induced by heat (path A) or CuO (path B) to form tert-butyl radical, which extracts hydrogen from silane to produce silicon center free radical. Instantaneous alkyl radical I is yielded by the addition of silyl free radical to 1. Then it is intercepted by heteroaryl groups in the molecule to obtain the intermediate II. The N-center free radical II causes the homolysis of the cyclic C-C bond and affords the keto radical III. The desired product 2 is eventually formed by the subsequent oxidation and deprotonation of III.

|
Download:
|
| Scheme 17. Mechanism of heteroarylsilylation of unactivated olefins. | |
{kind=link}
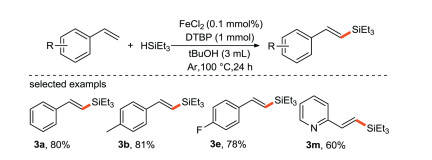
Our group [40] reported the selective synthesis of (E)-specific alkenyl silane using DTBP as an oxidant by iron-catalyzed cross-coupling reaction of silane with alkenes (Scheme 18). To the best of our knowledge, this is the first time that iron salts are used as electron donors to promote the formation of silicon center free radical.

|
Download:
|
| Scheme 18. Synthesis of alkenyl silane by cross-coupling of styrene and its derivatives. | |
{kind=link}
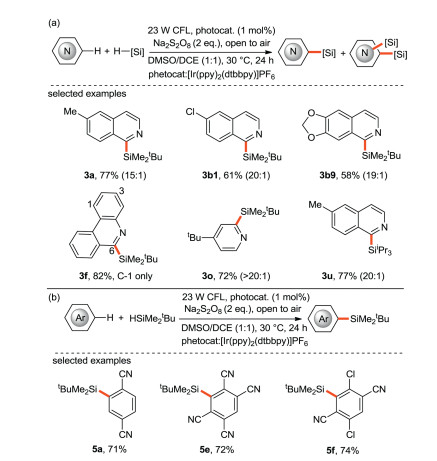
Li, Zhang et al. [41] reported a unique visible light-promoted photocatalytic silylation of C-H bond, which could directly couple trialkylhydrosilane with electron-deficient and electron-rich heteroaromatics (Scheme 19a) and cyano-substituted aromatics (Scheme 19b) with moderate to high yields and good regioselectivity. The protocol features operational simplicity, mild reaction conditions, and the use of safe and readily available Na2S2O8 as the radical initiators. Notably, the challenging bulky and inert trialkylhydrosilanes, such as (t-butyldimethyl)silane (tBuMe2SiH) and (triisopropyl)silane (iPr3SiH), also work smoothly in the system.

|
Download:
|
| Scheme 19. Cross-coupling reaction of hydrosilane with heteroaromatics or cyano-substituted aromatics. | |
{kind=link}
Wang and Quan et al. [42] demonstrated the copper-catalyzed stereospecific denitrosilylation of β-nitroolefins with silanes (Scheme 20). A series of (E)-specific vinylsilanes are synthesized in good yields, which can tolerate a variety of functional groups. It also proposes the advantages of using inexpensive and easily available substrates. The preliminary mechanistic studies disclose that the reaction may proceed via a radical pathway.

|
Download:
|
| Scheme 20. Denitrosilylation of β-nitroolefin. | |
{kind=link}
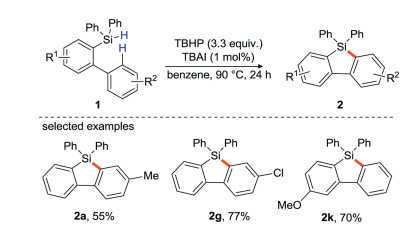
In addition to transition metals as electron donors, Studer's group [43] found that tetrabutylammonium iodide (TBAI) could also be used as an electron donor to initiate peroxide decomposition. They synthesize 9-silafluorenes from biaryldiphenylsilane using TBHP as a radical initiator, TBAI as an electron donor (Scheme 21). This strategy is carried out through intramolecular base-promoted homolytic aromatic substitution (BHAS). The author emphasize the concept of "electron is a catalyst" because these cyclizations are catalyzed by the electron [44].

|
Download:
|
| Scheme 21. Synthesis of 9-silafluorene by biaryldiphenylsilane intramolecular cyclization. | |
{kind=link}
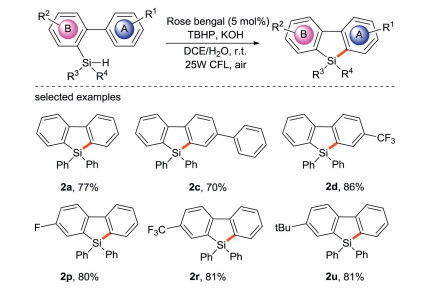
Furthermore, photocatalysts can sometimes provide electrons to decompose peroxides, thereby realizing radical silylation. In 2018, Jiang's group [45] reported the free radical silylation of biaryl hydrosilane induced by visible light for the synthesis of diphenylsilane (Scheme 22), which is the first photocatalytic synthesis of diphenylsilane. Satisfactory yields are obtained under mild and water/air compatible conditions using cheap organic dye photocatalysts, and a wide range of functional groups and substrates can be tolerated, so it is an effective and practical method for the synthesis of bifunctional silicon. Light/dark experiments and quantum yield measurement results support that there is a photocatalytic pathway rather than the chain process in the transformation.

|
Download:
|
| Scheme 22. Visible light-induced synthesis of diphenylsilane from biarylhydrosilane. | |
{kind=link}
4. Alkali metal tert-butoxides initiated radical silylation
Alkali metal tert-butoxides (KOtBu, NaO tBu) as strong bases play key roles in numerous organic transformations. More recently, several reports address that these bases can directly trigger the electron transfer processes or indirectly initiate the radical route after the tert-butoxide reacts with a solvent or additive [46-49].
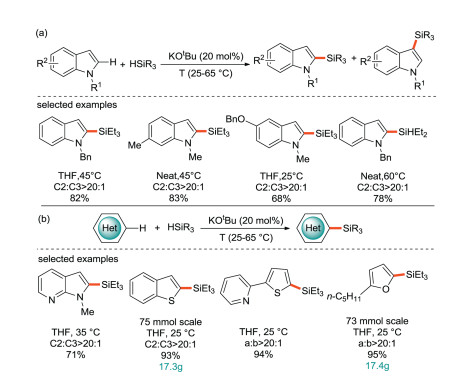
In 2015, the Grubbs' group reported on the silylation of C(sp2)-H bonds in aromatics catalyzed by KOtBu [50]. They have also mentioned a similar catalytic method before [51, 52]. It is found that potassium tert-butoxide can catalyze the direct silylation of aromatics with hydrosilane (Scheme 23a). The silylation is carried out under mild conditions without hydrogen acceptor, ligand or additive, and can be extended to more than 100 g scale under solvent-free conditions. At the same time, this C-H silylation method are compatible with a range of functional groups including pyridine, piperidine and furan (Scheme 23b), making it suitable for medicinal chemistry and the synthesis of alkaloid natural products.

|
Download:
|
| Scheme 23. KOtBu-catalyzed silylation of sp2 and sp3 CH bonds. | |
{kind=link}
In order to explore the mechanism of alkali-catalyzed silylation, several attempts have been made by researchers. Zare and coworkers [53] proposed an ionic mechanisms for the reaction. This mechanism is consistent with the ion intermediates detected in the experiment. The key step of the route is the nucleophilic attack of KOtBu on silane to form reactive penta-coordinated silicide, which limits the isomerization of Si-H. Nevertheless, this mechanism cannot explain the existence of free radicals during the reaction.
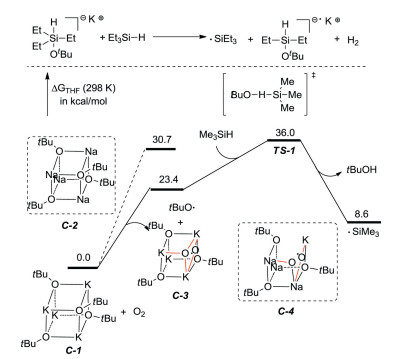
Grubbs research group [54] supposed a radical chain mechanism for the reaction based on experiments and computer theoretical research. A trialkylsilyl radical may be initially generated by homolytic cleavage of the Si-H bond of a hypercoordinated silicon species. Meanwhile, another route for the generation of this radical is also raised, in which traces of oxygen in the reaction system can react with [KOtBu]4 to generate a reactive peroxide, which can afford silicon radical by homolytic cleavage and hydrogen atom transfer. This route is also confirmed by DFT calculations (Scheme 24). The free radical clock and KIE experiments support the mechanism that the C-Si bond is added to the heterocyclic ring by silyl radical, and then β-hydrogen breaks down to form the product. DFT results reveal a reasonable energy distribution of the free radical mechanism and support the regioselectivity observed experimentally.

|
Download:
|
| Scheme 24. Possible radical initiation and free energy profile for generation of silyl radical triggered by traces of oxygen. | |
{kind=link}
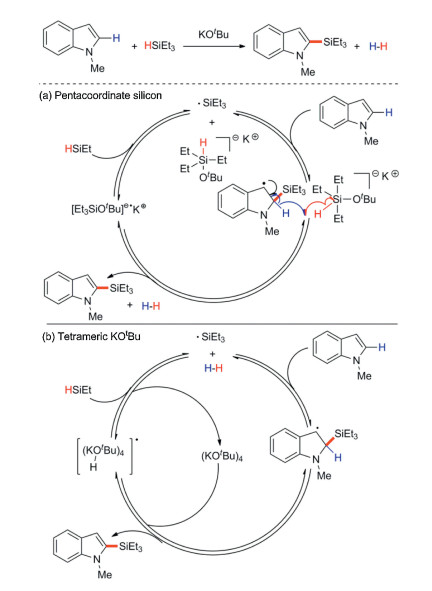
There are two tentative mechanisms: radical pathway mediated by pentacoordinate silicon (Scheme 25a) and tetrameric KOtBu (Scheme 25b). (1) The free radical process mediated by penta-coordinated silicon: A stable benzyl radical is formed by the addition of Et3Si· to indole at C2 position, and the β-H scission of the radical intermediate restores aromaticity in the indole system and generates H2, which provides entropy driving force for the whole reaction. Then, the silicate radical anion can react with the same amount of triethyl silane to regenerate Et3Si· and continue the chain process (Scheme 25a). (2) The free radical pathway mediated by tetramer KOtBu ([KOtBu]4): Silicified product and alkali-hydrogen radical adduct are produced by extracting hydrogen atoms from benzyl groups. Then, this free radical adduct reacts with hydrosilane to produce H2 and regenerates silyl radicals, thus completing the catalytic cycle (Scheme 25b).

|
Download:
|
| Scheme 25. Reaction mechanism of KOBu catalyzed silylation. | |
{kind=link}
5. Photocatalytically initiated radical silylation
Organic photochemical synthesis has developed rapidly in the past few decades [55-60], in which single electron transfer (SET), energy transfer and hydrogen atom transfer (HAT) greatly increase the chance of activation of C-H bonds. At present, more and more researchers are devoted to investigate photocatalytic free radical pathways to synthesize organosilicons. There are two pathways for photocatalytically initiated radical silylation: (I) The photocatalyst provides an electron to decompose the peroxide which has been addressed in Section 3; (II) the photocatalyst obtains electron from HAT catalyst or Br- to generate radical species. Herein, we summarize the latest progresses on photocatalytically initiated radical silylation via the pathway II.
The group of Wu [61] has done several excellent researches in the field of photocatalytic silylation. In 2017, they developed the first visible light-driven metal-free hydrosilylation of electron-deficient and electron-rich olefins through photoredox and HAT catalysis (Scheme 26). The hydrosilylation of electron-deficient olefins is realized by the synergistic combination of organic photoredox catalyst 4CzIPN and quinuclidin-3-ly acetate, meanwhile the hydrosilylation of electron-rich olefins is achieved by the combination of photoredox catalysis and polarity reversal catalysis. This protocol has the characteristics of atomic and redox economy, mild conditions and a wide range of substrates, and it is easy to realize large-scale production. Therefore, it is possible to apply this strategy for further synthesizing functionalized silicon-containing molecules.

|
Download:
|
| Scheme 26. Visible-light mediated metal-free hydrosilation of olefins. | |
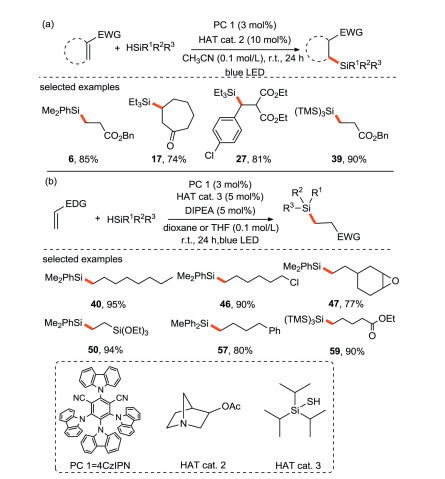{kind=link}
In 2018, the same group realized the visible light-mediated metal-free bifunctional assembly of olefins with CO2 and hydrosilane to synthesize β-silacarboxylic acid through the combination of photocatalysis and hydrogen atom transfer [62] (Scheme 27). This bifunctionalization reaction has the advantages of step and redox economy, mild reaction conditions, free of external oxidant and a wide range of substrates, which is easy to be scaled up through continuous-flow technology. It is worth mentioning that this study activates olefins through a redox-neutral of Si-H or C–H activation instead of oxidative fashion, which provides a new strategy for the realization of olefin bifunctionalization using abundant and cheap chemical raw materials.

|
Download:
|
| Scheme 27. Visible-light mediated bifunctionalization of olefin with CO2 and hydrosilane. | |
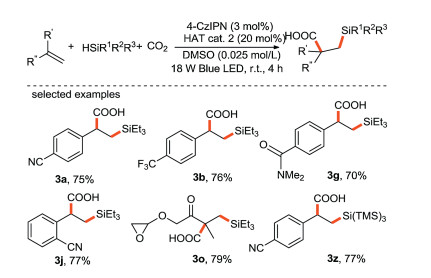{kind=link}
Wei and Sortais et al. [63] reported that direct reduction of various carboxylic acids to disilyl ethers was effectively catalyzed by Re under mild conditions (room temperature, irradiation 350 or 395 nm) (Scheme 28). Aliphatic carboxylic acid can be easily converted into the corresponding disilyl ether with lower catalyst loading (0.5 mol%). In addition, this new approach is applied to a series of benzoic acid derivatives which are difficult to be reduced by traditional methods.

|
Download:
|
| Scheme 28. Photocatalytic reduction of carboxylic acid to disilyl ether. | |
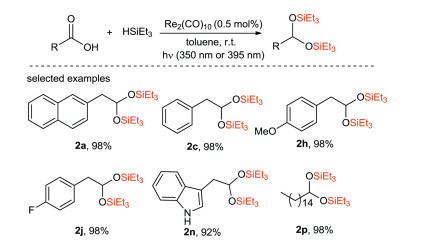{kind=link}
Uchiyama and Wang et al. [64] reported first approach for the generation of silyl radicals from silyl carboxylic acid: photo-induced decarboxylation of silacarboxylic acid catalyzed by 4CzIPN. Silyl radicals are obtained by hydrogen atom transfer, electron transfer and de-CO2 reaction, then they react with olefins to obtain a wide range of adducts with high yields (Scheme 29a). In addition, the experimental results show that D2O can be used as a deuterium source to obtain deuterated silylated products (Scheme 29b), suggesting that water is the main hydrogen source.

|
Download:
|
| Scheme 29. Photo-induced radical silylation reaction from silyl carboxylic acid. | |
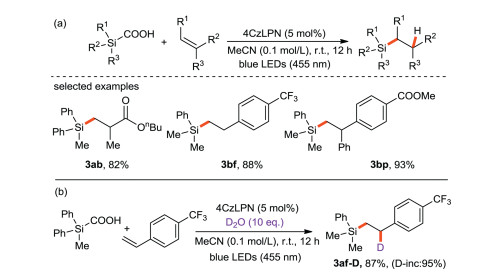{kind=link}
Zhang and Hu [65] reported Ni-catalyzed arylsilylation of electron-deficient terminal olefins using aryl bromides and TMS3SiH as reaction partners (Scheme 30). The silicon center radical is generated by photoredox, and the arylation proceeded via Ni-catalyzed cross coupling.

|
Download:
|
| Scheme 30. Photo-induced arylsilylation of alkenes. | |
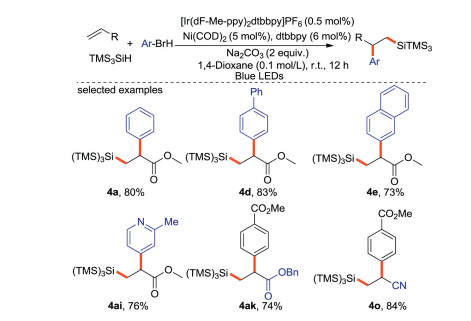{kind=link}
The mechanism of arylsilylation reaction was proposed (Scheme 31). Ni0 catalyst A is oxidized by bromobenzene to form NiII intermediate B, and Ir(III) is excited by luminescence to react with Br- to produce bromine radical by single electron tranfer. Bromine radical extracts hydrogen from silane to form silicon center free radicals E. The silylated alkyl radical intermediate F is formed by adding E to the substrate 1a. F is captured by NiII intermediate B to obtain NiIII intermediate C, which is reduced and eliminated by C-C bond to form aryl silylation product 4a. At the same time, the NiI complex is reduced by Ir(II) to regenerate the Ni0 complex and Ir(III).

|
Download:
|
| Scheme 31. Catalytic cycle of photo-induced silylation of terminal olefins. | |
{kind=link}
6. Alkyl radical initiated silylation
Although a large number of radical silylation has been developed for the formation of C(sp2)-Si and C(sp)-Si bonds, the construction of C(sp3)-Si bonds by radical pathway is rare because it is difficult to directly activate C(sp3)-H or C(sp3)-X bonds by silyl radicals. To achieve this goal, several alkyl radical initiated silylation have been disclosed. In this type of silylation, no silicon center radicals were detected under peroxide-free conditions, and the homolytic of active C(sp3)-X bonds can be realized by copper catalysts to produce alkyl free radicals which can react with Si-B reagent to construction of C(sp3)-Si bonds.
The research group of Oestreich [66] introduced a copper-catalyzed silylation with alkyl iodide using a Si-B reagent as silicon pronucleophile (Scheme 32a). In some cases of tethered alkene, a 5-exo-trig radical cyclization occurs before the generation of C(sp3)-Si bonds. (Scheme 32b). This protocol provides moderate to excellent yield of alkyl silanes (42%–99%) with a wide range of substrate scope. The experimental exploration and quantum-chemical calculation show that the mechanism includes a free radical process, in which alkyl radical is the key intermediate to trigger the reaction. A complete mechanism is proposed in Scheme 33.

|
Download:
|
| Scheme 32. Silylation of alkyl iodides with silicon pronucleophiles. | |
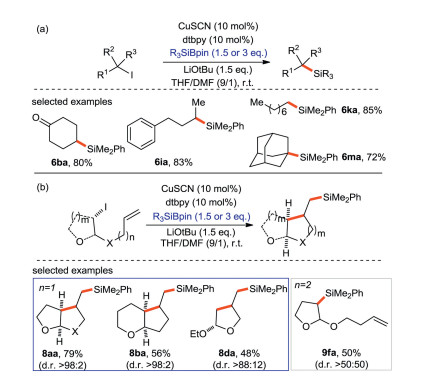{kind=link}

|
Download:
|
| Scheme 33. Tentitive catalytic cycle of the copper-catalyzed C(sp3)-Si coupling of alkyl iodide and Si-B reagent. | |
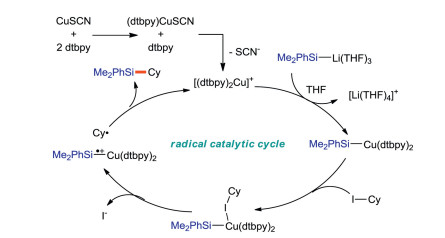{kind=link}
In 2017, Oestreich and Xue [67] reported the decarboxylsilylation of aliphatic N-hydroxyphthalimide esters (NHPI) with Si-B silicon pronucleophiles catalyzed by CuTc (copper(I) thiophene-2-carboxylate). This approach follows the free radical mechanism. The primary and secondary alkyl groups couple effectively (Scheme 34a), while the tertiary alkyl groups only afford a poor yield (7ma) owing to the sterically hindered effects. α-Nitrogen substituted substrates can afford higher yields of corresponding products because lone pair electrons of N can stable the alkyl radicals (Scheme 34b).

|
Download:
|
| Scheme 34. Decarboxylation silylation of aliphatic N-hydroxyphthalimide (NHPI) ester with Si-B reagent. | |
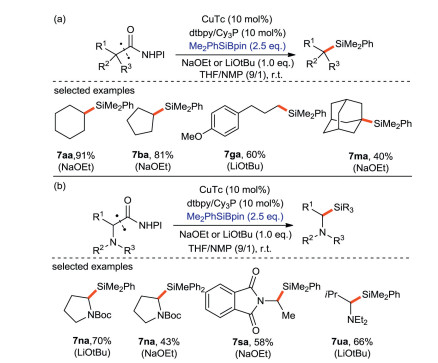{kind=link}
In 2018, the same group have reported a new copper catalyzed silylation [68] for the synthesis of 1, 1-disilylated alkanes from alkyl dibromides and silicon pronucleophiles (Scheme 35a). According to the previous work experience, the different alkyl electrophile may make Cu-catalyzed C-Si cross-coupling follow different mechanisms. In order to insight into the mechanism, the authors perform several free radical clock experiment. These results indicate that the bonding mechanisms of the two C(sp3)-Si bonds are different. α-Bromo-substituted alkyl bromide reacts through the ionic pathway and the intermediate α-silylated alkyl bromide couples through the radical pathway.

|
Download:
|
| Scheme 35. The copper-catalyzed silylation for the synthesis of 1, 1-disilylated alkanes from alkyl dibromides and its mechanism. | |
{kind=link}
According to experimental and calculation results, a possible mechanism is proposed in Scheme 35b. First, the copper-based silicon nucleophile 10 is obtained from 8 and 9, then 10 replaces one bromine on the substrate to afford brominated silane 7. The unactivated 7 reacts with 10 generates free radical cation 11 and alkyl radical 12 by single electron transfer. Finally, 12 and 11 are compounded to form 1, 1-disilylated alkane 2.
Cheng and Mankad [69] have reported for the first time that Cu(I) catalyzed one-pot carbonylative silylation of unactivated alkyl halides with CO and silicon pronucleophiles, and alkyl substituted acylsilanes were synthesized with high yields (Scheme 36a). This protocol is suitable for primary, secondary and tertiary alkanes, and can tolerate a variety of functional groups. On the basis of the existing results and previous work conclusions, the catalytic mechanism of this reaction is proposed (Scheme 36b). Firstly, NaOPh assisted Cu(I) and Me2PhSiBpin to form silicon-based copper(I) complex A. Then, a single electron transfer (SET) occurs between A and alkyl halides to produce silylcopper (II) complex B and alkyl radical R·, which is carbonylated with CO to form acyl radical C. B couples with C to yield Cu(III) complex intermediate D. Finally, the product acylsilane was obtained by reduction and elimination of D, and the Cu(I) was regenerated to enter the next catalytic cycle.

|
Download:
|
| Scheme 36. The carbonyl silylation reaction and its mechanism. | |
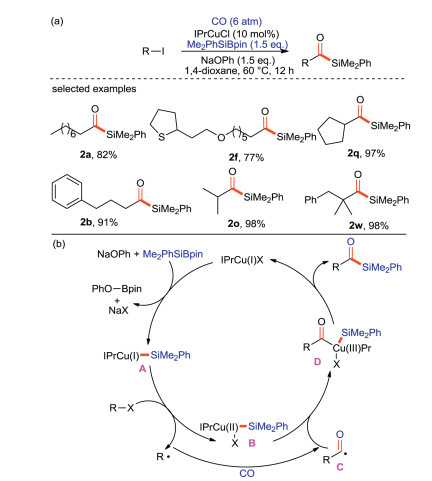{kind=link}
7. Other ways initiated radical silylation
In most of strategies, stoichiometric peroxides are necessary as the initiator of silyl radicals and the H acceptor. The formation of silyl radicals can be achieved through photoredox and HAT catalysis in the absence of peroxides, but the use of expensive photocatalysts is norm [61, 62, 65]. To eliminate the use of peroxides, other ways for the formation of silyl radicals have been disclosed.
Kegnæs et al. [70] prepared a new heterogeneous catalyst Ni1Co1@NC for silylation of aromatic aldehydes (Scheme 37a), by which a series of silylated pinacol coupling products are synthesized. The reaction mechanism is different from the classical pinacol coupling pathway, in which the heterogeneous catalyst facilitates easy access to silyl radicals, thereby circumventing the usual need for explosive initiators to access these species.

|
Download:
|
| Scheme 37. Silylative pinacol coupling reaction between aromatic aldehyde and hydrosilane and its mechanism. | |
{kind=link}
As shown in Scheme 37b, Ni1Co1@NC first cleaved HSiEt3, to release silicon center free radicals, and silicon center free radicals attack the oxygen atoms of aldehydes to form stable benzyl radicals. There are two ways to form silylated pinacol coupling products. One is that benzyl radical is added to benzaldehyde to form alkoxy radical which may be promoted by PhCO2H, and then combined with silicon central radical to obtain the coupling product. Alternatively, two benzyl radicals are dimerized to yield a coupling product directly. Finally, a pressure buildup in the reaction vessel is observed owing to the release of H2 during the reaction.
Oswald and Woerpel reported [71] a intramolecular silyl peroxidation of unsaturated diisopropylsilyl ethers catalyzed by Co(thd)2 (thd=2, 2, 6, 6-tetramethyl-3, 5-heptanedionato) to form 3-sila-1, 2, 4-trioxepanes internal peroxides. A more stable hexagonal diisopropylsilane acetal can be obtained by PPh3 reduction (Scheme 38a). The synthetic mechanism of 3-sila-1, 2, 4-trioxepanes is also studied in this paper. The experimental results indicate that HSiEt3 participates in the formation of CoIIIH species (Scheme 38b). First, tBuOOH and HSiEt3 trigger the formation of CoIIIH, which add to the double bond of unsaturated diisopropylsilyl ethers molecule I to afford II. CoII was dissociated from II to obtain alkyl radical III. III are peroxidized to obtain peroxide radical IV. Then, CoII provides a single electron to combine with IV to form the CoIII-peroxy intermediate V. V has two possible reaction pathways: (1) A transition state VI is generated by the intramolecular metal transfer reaction, then VI produces 3-sila-1, 2, 4-trioxepane VIII by removing CoIIIH; (2) metal transfer reaction occurs between V and HSiEt3, in which CoIIIH is eliminated to form linear methylsilane peroxide VII.

|
Download:
|
| Scheme 38. Intramolecular silicon-based peroxidation reaction and its mechanism. | |
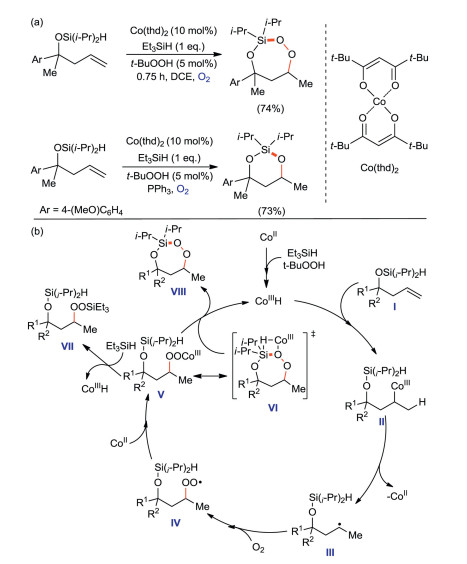{kind=link}
8. Summary and outlook
The article mainly concentrates on the research progress of the organosilicide synthesis based on free radical pathways since 2016. According to the different initiation mechanism of free radicals, they can be divided into the following categories: 1) peroxide initiated radical silylation; 2) transition metal induced peroxide decomposition initiated radical silylation; 3) alkali initiated radical silylation; 4) photocatalytically initiated radical silylation; 5) alkyl radical initiated silylation.
At present, the radical strategies for the synthesis of organosilicide initiated by electron-induced peroxides decomposition or peroxides are more mature, but most of the studies require of stoichiometric peroxides as the initiator of silyl radicals and the H acceptor, and the recycling and reuse of transition metal catalysts still unresolved. The formation of silyl radicals can be achieved through combination of photoredox and HAT catalysis under peroxide-free conditions, but the expensive photocatalysts are norm. Although several alkyl radical initiated silylations have been developed for the construction of C(sp3)-Si bonds, there is still a lot of room to expand the reaction types and improve conditions of this strategy.
To solve these issues above, it is necessary and significant to explore new and sustainable strategies or catalysts for the radical silylation. For example, (1) the recycling and reuse of transition metal catalysts can be implemented by the employ of non-noble metal based heterogeneous catalysts; (2) the combination of single electron transfer and acceptorless dehydrogenation catalyzed by transition metal or the use of molecular oxygen may be an alternative to the use of peroxides; (3) new strategies for accessing silyl or alkyl radicals, has a high potential to expand the reaction types or improve reaction conditions.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge the Fundamental Research Funds for the Central Universities (No. 30920021120), Key Laboratory of Biomass Energy and Material, Jiangsu Province (No. JSBEM201912), the National Natural Science Foundation of China (No. 21905089) and the Chinese Postdoctoral Science Foundation (No. 2019M662775) for financial support.
| [1] |
M.A. Brook, Silicon in Organic, Organometallic and Polymer Chemistry, John Wiley & Sons Ltd., Chichester, 2000.
|
| [2] |
K.L. Lee, Angew. Chem. Int. Ed. 56 (2017) 3665-3669. DOI:10.1002/anie.201612460 |
| [3] |
A.K. Franz, S.O. Wilson, J. Med. Chem. 56 (2013) 388-405. DOI:10.1021/jm3010114 |
| [4] |
A.G. Schafer, J.M. Wieting, A.E. Mattson, Org. Lett. 13 (2011) 5228-5231. DOI:10.1021/ol2021115 |
| [5] |
A.M. Hardman-Baldwin, A.E. Mattson, ChemSusChem 7 (2014) 3275-3278. DOI:10.1002/cssc.201402783 |
| [6] |
J.M. Wieting, T.J. Fisher, A.G. Schafer, M.D. Visco, Eur. J. Org. Chem. 3 (2015) 525-533. |
| [7] |
J. Hu, A.X. Zhang, L. Chen, W.Q. Li, X.H. Zeng, Silicone Mater. 33 (2019) 218-246. |
| [8] |
S.E. Denmark, C.S. Regens, Acc. Chem. Res. 41 (2008) 1486-1499. DOI:10.1021/ar800037p |
| [9] |
C. Chatgilialoglu, Acc. Chem. Res. 25 (1992) 188-194. DOI:10.1021/ar00016a003 |
| [10] |
A.E. Portera, N. Patela, J.N. Skepperb, S.M. Besta, W. Bonfielda, Biomaterials 25 (2004) 3303-3314. DOI:10.1016/j.biomaterials.2003.10.006 |
| [11] |
R. Jugdaohsingh, K.L. Tucker, N. Qiao, et al., J. Bone Miner. Res. 19 (2004) 296-307. |
| [12] |
W.X. Xu, H.L. Teng, Y. Luo, et al., Chem. Asian J. 15 (2020) 753-756. DOI:10.1002/asia.202000089 |
| [13] |
T. Komiyama, Y. Minami, T. Hiyama, ACS Catal. 7 (2017) 631-651. DOI:10.1021/acscatal.6b02374 |
| [14] |
J. Scharfbier, M. Oestreich, Synlett 27 (2016) 1274-1276. DOI:10.1055/s-0035-1561407 |
| [15] |
J. Jia, X.Q. Zeng, Z.L. Liu, et al., Org. Lett. 22 (2020) 2816-2821. DOI:10.1021/acs.orglett.0c00809 |
| [16] |
B. Raya, S. Jing, V. Balasanthiran, T.V. RajanBabu, ACS Catal. 7 (2017) 2275-2283. DOI:10.1021/acscatal.6b03373 |
| [17] |
L. Omann, M. Oestreich, Organometallics 36 (2017) 767-776. DOI:10.1021/acs.organomet.6b00801 |
| [18] |
Z.Y. Cheng, S.P. Xing, J. Guo, et al., Chin. J. Chem. 37 (2019) 457-461. DOI:10.1002/cjoc.201900079 |
| [19] |
H.B. Peng, J.T. Yu, Y. Jiang, J. Cheng, Org. Biomol. Chem. 13 (2015) 10299-10302. DOI:10.1039/C5OB01855B |
| [20] |
H. Qrareya, D. Dondi, D. Ravelli, M. Fagnoni, ChemCatChem 7 (2015) 3350-3357. DOI:10.1002/cctc.201500562 |
| [21] |
Z. Cao, Q. Zhua, Y.W. Lin, W.M. He, Chin. Chem. Lett. 30 (2019) 2132-2138. DOI:10.1016/j.cclet.2019.09.041 |
| [22] |
K.J. Liu, T.Y. Zeng, J.L. Zeng, et al., Chin. Chem. Lett. 30 (2019) 2304-2308. DOI:10.1016/j.cclet.2019.10.031 |
| [23] |
X.J. Shang, Z.Q. Liu, Org. Biomol. Chem. 14 (2016) 7829-7831. DOI:10.1039/C6OB00797J |
| [24] |
J.S. Li, J. Wu, ChemPhotoChem 2 (2018) 839-846. DOI:10.1002/cptc.201800110 |
| [25] |
H.T. Huang, T. Li, J.Z. Wang, G.P. Qin, T.B. Xiao, Chin. J. Org. Chem. 39 (2019) 1511-1521. DOI:10.6023/cjoc201903078 |
| [26] |
J.R. Wilkinson, C.E. Nuyen, T.S. Carpenter, S.R. Harruff, R.V. Hoveln, ACS Catal. 9 (2019) 8961-8979. DOI:10.1021/acscatal.9b02762 |
| [27] |
L. Xu, S. Zhang, P.F. Li, Org. Chem. Front. 2 (2015) 459-463. DOI:10.1039/C5QO00012B |
| [28] |
Y.M. Lin, G.P. Lu, R.K. Wang, W.B. Yi, Org. Lett. 19 (2017) 1100-1103. DOI:10.1021/acs.orglett.7b00126 |
| [29] |
R. Sakamoto, B.N. Nguyen, K. Maruoka, Asian J. Org. Chem. 7 (2018) 1085-1088. DOI:10.1002/ajoc.201800282 |
| [30] |
C. Zhang, J.X. Pi, L. Wang, P. Liu, P.P. Sun, Org. Biomol. Chem. 16 (2018) 9223-9229. DOI:10.1039/C8OB02670J |
| [31] |
X.H. Chang, Z.L. Wang, M. Zhao, et al., Org. Lett. 22 (2020) 1326-1330. DOI:10.1021/acs.orglett.9b04645 |
| [32] |
L.Z. Zhang, D. Liu, Z.Q. Liu, Org. Lett. 17 (2015) 2534-2537. DOI:10.1021/acs.orglett.5b01067 |
| [33] |
J. Gu, C. Cai, Chem. Commun. 52 (2016) 10779-10782. DOI:10.1039/C6CC05509E |
| [34] |
L.Z. Zhang, Z.J. Hang, Z.Q. Liu, Angew. Chem. Int. Ed. 55 (2016) 236-239. DOI:10.1002/anie.201509537 |
| [35] |
Y. Lan, X.H. Chang, P. Fan, et al., ACS Catal. 7 (2017) 7120-7125. DOI:10.1021/acscatal.7b02754 |
| [36] |
Z.F. Yan, J. Xie, C.J. Zhu, Adv. Synth. Catal. 359 (2017) 4153-4157. DOI:10.1002/adsc.201700926 |
| [37] |
Y. Yang, R.J. Song, X.H. Ouyang, et al., Angew. Chem. Int. Ed. 56 (2017) 7916-7919. DOI:10.1002/anie.201702349 |
| [38] |
Z.B. Xu, L. Chai, Z.Q. Liu, Org. Lett. 19 (2017) 5573-5576. DOI:10.1021/acs.orglett.7b02717 |
| [39] |
H. Zhang, X.X. Wu, Q. Zhao, C. Zhu, Chem. Asian J. 13 (2018) 2453-2457. DOI:10.1002/asia.201800150 |
| [40] |
R. Xu, C. Cai, Catal. Commun. 107 (2018) 5-8. DOI:10.1016/j.catcom.2017.12.023 |
| [41] |
S.H. Liu, P. Pan, H.Q. Fan, et al., Chem. Sci. 10 (2019) 3817-3825. DOI:10.1039/C9SC00046A |
| [42] |
X. Zhang, M.X. Liu, T.L. Wang, et al., Org. Chem. Front. 6 (2019) 3365-3368. DOI:10.1039/C9QO00819E |
| [43] |
D. Leifert, A. Studer, Org. Lett. 17 (2015) 386-389. DOI:10.1021/ol503574k |
| [44] |
A. Studer, D.P. Curran, Nat. Chem. 6 (2014) 765-773. DOI:10.1038/nchem.2031 |
| [45] |
C. Yang, J. Wang, J.H. Li, et al., Adv. Synth. Catal. 360 (2018) 3049-3054. DOI:10.1002/adsc.201800417 |
| [46] |
J.P. Barham, G. Coulthard, T. McGuire, T. Tuttle, J.A. Murphy, J. Am. Chem. Soc. 138 (2016) 7402-7410. DOI:10.1021/jacs.6b03282 |
| [47] |
M. Rueping, M. Leiendecker, A. Das, T. Poisson, L. Bui, Chem. Commun. 47 (2011) 10629-10631. DOI:10.1039/c1cc14297f |
| [48] |
E. Shirakawa, T. Hayashi, Chem. Lett. 41 (2012) 130-134. DOI:10.1246/cl.2012.130 |
| [49] |
E. Doni, S.Z. Zhou, J.A. Murphy, Molecules 20 (2015) 1755-1774. DOI:10.3390/molecules20021755 |
| [50] |
A.A. Toutov, W.B. Liu, K.N. Betz, et al., Nature 518 (2015) 80-84. DOI:10.1038/nature14126 |
| [51] |
A. Fedorov, A.A. Toutov, N.A. Swisher, R.H. Grubbs, Chem. Sci. 4 (2013) 1640-1645. DOI:10.1039/c3sc22256j |
| [52] |
A.A. Toutov, W.B. Liu, K.N. Betz, B.M. Stoltz, R.H. Grubbs, Nat. Protoc. 10 (2015) 1897-1903. DOI:10.1038/nprot.2015.118 |
| [53] |
S. Banerjee, Y.F. Yang, I.D. Jenkins, et al., J. Am. Chem. Soc. 139 (2017) 6880-6887. DOI:10.1021/jacs.6b13032 |
| [54] |
W.B. Liu, D.P. Schuman, Y.F. Yang, et al., J. Am. Chem. Soc. 139 (2017) 6867-6879. DOI:10.1021/jacs.6b13031 |
| [55] |
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075-10166. DOI:10.1021/acs.chemrev.6b00057 |
| [56] |
R. Brimioulle, D. Lenhart, M.M. Maturi, T. Bach, Angew. Chem. Int. Ed. 54 (2015) 3872-3890. DOI:10.1002/anie.201411409 |
| [57] |
K.L. Skubi, T.R. Blum, T.P. Yoon, Chem. Rev. 116 (2016) 10035-10074. DOI:10.1021/acs.chemrev.6b00018 |
| [58] |
J. Xuan, W.J. Xiao, Angew. Chem. Int. Ed. 51 (2012) 6828-6838. DOI:10.1002/anie.201200223 |
| [59] |
L.Y. Xie, T.G. Fang, J.X. Tan, et al., Green Chem. 21 (2019) 3858-3863. DOI:10.1039/C9GC01175G |
| [60] |
L.L. Wang, M. Zhang, Y.L. Zhang, et al., Chin. Chem. Lett. 31 (2020) 67-70. DOI:10.1016/j.cclet.2019.05.041 |
| [61] |
R. Zhou, Y.Y. Goh, H.W. Liu, et al., Angew. Chem. Int. Ed. 56 (2017) 16621-16625. DOI:10.1002/anie.201711250 |
| [62] |
J. Hou, A. Ee, H. Cao, H.W. Ong, J.H. Xu, J. Wu, Angew. Chem. Int. Ed. 57 (2018) 17220-17224. DOI:10.1002/anie.201811266 |
| [63] |
D. Wei, R. Buhaibeh, Y. Canac, J.B. Sortais, Org. Lett. 21 (2019) 7713-7716. DOI:10.1021/acs.orglett.9b02449 |
| [64] |
N.X. Xu, B.X. Li, C. Wang, M. Uchiyama, Angew. Chem. Int. Ed. 59 (2020) 2-8. DOI:10.1002/anie.201914768 |
| [65] |
Z.K. Zhang, X.L. Hu, ACS Catal. 10 (2020) 777-782. DOI:10.1021/acscatal.9b04916 |
| [66] |
W.C. Xue, Z.W. Qu, S. Grimme, M. Oestreich, J. Am. Chem. Soc. 138 (2016) 14222-14225. DOI:10.1021/jacs.6b09596 |
| [67] |
W.C. Xue, M. Oestreich, Angew. Chem. Int. Ed. 56 (2017) 11649-11652. DOI:10.1002/anie.201706611 |
| [68] |
H. Hazrati, M. Oestreich, Org. Lett. 20 (2018) 5367-5369. DOI:10.1021/acs.orglett.8b02281 |
| [69] |
L.J. Cheng, N.P. Mankad, J. Am. Chem. Soc. 142 (2020) 80-84. DOI:10.1021/jacs.9b12043 |
| [70] |
S. Kramer, F. Hejjo, K.H. Rasmussen, S. Kegnæs, ACS Catal. 8 (2018) 754-759. DOI:10.1021/acscatal.7b02788 |
| [71] |
J.P. Oswald, K.A. Woerpel, J. Org. Chem. 84 (2019) 7564-7574. DOI:10.1021/acs.joc.9b00642 |