 2021, Vol. 32
2021, Vol. 32 


b Department of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK
Asymmetric hydrogenation of unsaturated compounds with molecular hydrogen is one of the most important chemical processes, which have been applied to producing important chiral intermediates for pharmaceuticals, fragrances and agrochemicals [1]. Generally, expensive noble metal catalysts, containing Ru, Rh, Pd and Ir metals, are employed in order to obtain high activity and enantioselectivity for asymmetric hydrogenation. Different from those noble-metal catalysts, some chiral catalysts containing earth-abundant metals, such as iron, cobalt, nickel and manganese, have attracted much attention for asymmetric hydrogenation and transfer hydrogenation, due to their economic and environmental benefits [2, 3]. Amongst these catalysts, cobalt catalysts are promising candidates for the hydrogenation of unsaturated substrates. In fact, some cobalt complexes were used as catalysts for hydrogenation reactions earlier [4]. However, harsh conditions, such as high temperature and high pressure, are generally required, limiting their practical usefulness. Recently, some well-defined cobalt catalysts have been reported for successful hydrogenation of unsaturated substrates containing C=C [5], C=O [3f, 6] and C=N [3e, 7] bonds under mild conditions. The cobalt-catalyzed asymmetric hydrogenation of unsaturated C=C double bonds is particularly impressive, giving highly enantioselectivities and conversions, sometimes better than those achieved with noble metal catalysts. However, the cobalt-catalyzed hydrogenation of the polar C=O bonds is less developed. To the best of our knowledge, there are only five examples of cobalt-catalyzed homogenous hydrogenation of ketones under mild conditions, and only one of them is enantioselective (Scheme 1). In 2012, Hanson and co-workers reported an example cobalt-catalytic hydrogenation of C=C, C=O, C=N bonds using a PNP-cobalt(Ⅱ)-alkyl catalyst 1, with a substrate to catalyst ratio of 50:1 [6h]. In 2014, Wolf, von Wangelin and co-workers developed an arenecobalt catalyst 2; a high catalyst loading (5%) was required to obtain high yields [6e]. In 2015, Kempe and co-workers found that a triazine-based cobalt-PNP catalyst 3 can affect the hydrogenation of ketones at a lower catalyst loading (down to 0.25%), affording excellent yields [6d]. More recently, Liu and co-workers reported a highly active phosphine-free NHC-Co(Ⅱ) catalyst 4 for the hydrogenation of ketones, affording excellent yields with only 0.01% catalyst loading [6a]. Despite the efforts, the cobalt-catalyzed asymmetric hydrogenation of ketones remains rare. A breakthrough was reported by Li and co-workers in 2016 [3f]. Using the chiral PNNP-cobalt catalyst 5 (2mol%), moderate to good enantioselectivities for asymmetric hydrogenation of ketones were obtained. Herein, we report a new catalytic system for asymmetric hydrogenation of ketones with a chiral PNN-Co catalyst 6 [8] in combination with an achiral mono-phosphine ligand. Notably, the achiral mono-phosphine ligand plays an important role for the stereoselectivity, improving the enantioselectivity from 3%ee to 85% ee for the model reaction.

|
Download:
|
| Scheme 1. Cobalt catalysts for the hydrogenation of C=O bonds. | |
A series of chiral PNN ligands [9] with varying electronic and steric properties were designed and synthesized (Scheme 2). Treatment of the ligands La-Lg with CoCl2 in THF led to the cobalt complexes 6a-6g in good yields. The structure of 6g was confirmed by X-ray crystallographic analysis (CCDC: 1997410).

|
Download:
|
| Scheme 2. The synthesis of cobalt catalysts. | |
The spectra of the electron paramagnetic resonance indicate that 6g is a pentacoordinated low-spin cobalt(Ⅱ) complex (details in Supporting information).
Having obtained these cobalt catalysts, we turned our attention to examining the hydrogenation of acetophenone with 6a as the catalyst firstly. The reduction was carried out under similar reaction conditions as reported by Kempe [6d]. As showed in Table 1, no reaction was observed with the cobalt(Ⅱ) catalyst under such conditions (entry 1). Activating the catalyst with two equivalents of NaHBEt3 led to a full conversion for the hydrogenation reaction, but unfortunately, almost a racemic product was obtained (entry 2). Considering the activated, 16e− catalyst can coordinate a solvent molecule or another 2-electron donor easily on its vacant site, it would be possible to coordinate an electron-rich phosphine ligand, which may alter the chiral environment. With this hypothesis, tributylphosphane was chosen as an additive firstly. The catalytic result demonstrated that the addition of tributylphosphane indeed affected the enantioselectivity (entry 3). And increasing the amount of additive led to improvement of enantioselectivities, although the catalytic activities eroded slightly (entries 3–5). Increasing the amount of base to 20% led to a full conversion but decreasing its enantioselectivity from 30% to 5% (Table 1, entry 6). Following these results, we then examined some kinds of stronger or weaker base under the optimized conditions (Table S1 in Supporting information), and found that Cs2CO3 gave the best results with full conversion and 39% enantioselectivity (entry 7). Next, other cobalt catalysts containing different substitutions were examined (entries 9–14).
|
|
Table 1 Optimization of reaction conditions for cobalt-catalyzed hydrogenation of acetophenone 1aa.a |

{kind=link}
{kind=link}
The best one is the catalyst 6g, affording full conversion and 43% enantioselectivity (entry 14). After the evaluation of several solvents (Table S2 in Supporting information), Et2O was chosen as the solvent, which gave the product with full conversion and 50% enantioselectivity (entry 14). Decreasing hydrogen pressure or temperature resulted in a similar enantioselectivity but lower conversion (Table S2). Since the phosphine ligand as the additive was found to exert a significant effect on the enantioselectivity (Table 1, entries 2 vs. 4, 3% vs. 30%), a series of phosphines were then subjected to the model reaction and some notable examples are summarized in Table 1 (see Fig S1 in Supporting information for more details). As expected, the phosphine additives significantly influenced the stereochemical outcome of the reaction, giving products with enantioselectivities ranging from 38% to 85% and good to excellent conversions. As can be seen from Table 1, the additives based on tri(2-furyl) phosphine (L2) and its derivatives (L3-L8) afforded much better enantioselectivities than others. Particularly, the bulky electron-donating ethyl substitute on the furyl group (L6) gave the best results in terms of both conversion and enantioselectivity. In contrast, the electron-withdrawing substituent on the furyl group (L7) or fused aromatic ring (L8) led to a remarkable decrease in conversion and enantioselectivity.
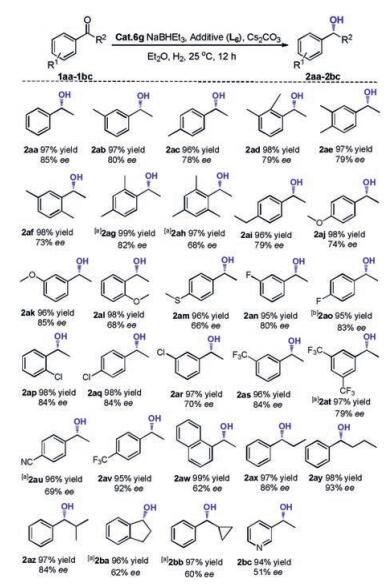
Having established workable conditions for the cobalt catalytic asymmetric hydrogenation of acetophenone, we subsequently turned to exploring the scope of aryl ketones. The results are listed in Scheme 3. As can be seen, all the substrates could undergo smooth hydrogenation under the identical reaction conditions, giving excellent isolated yields with moderate to good enantioselectivities. The enantioselectivity was relatively insensitive to the substitution on the phenyl ring (from 2aa to 2aw). Regardless of whether the substrate contains an electron-donating group or electron-withdrawing group (1aa-1av), the enantioselectivity was not very significantly affected, with the enantiomer excesses varying between 66% and 92%. Interestingly, for the substrate with a para-CF3 group (1av), a higher enantioselectivity 92% was achieved. Furthermore, the substrate with a sulfide group or 1-naphanthyl ketone could proceed smoothly with 60% ee (1am) and 62% ee (1aw). Changing the R2 substitution on the substrate led to a remarkable effect on the enantioselectivity (1ax-1az). Increasing the chain length of R2 alkyl group led to the improvement of enantioselectivity (2aa, 2ax-2by); for example, the substrate 1ay could be hydrogenated to 2ay with 93% enantioselectivity. However, in contrast to the linear alkyl groups, bulkier substitutes did not benefit the eantioselectivities (2az-2bb). Finally, the meta-acetyl pyridine could also be hydrogenated fully with 51% enantioselectivity (2bc).

|
Download:
|
| Scheme 3. Scope of substrates. Reaction conditions: Acetophenone (0.25 mmol), 6g (0.005 mmol), NaBHEt3 (0.01 mmol), Cs2CO3 (0.0125 mmol), L6 (0.0075 mmol), Et2O (0.500 mL), 25 ℃, H2 (40 bar), 12 h. The conversions and ee values were determined by GC analysis. aThe reaction time was 20 h. | |
{kind=link}
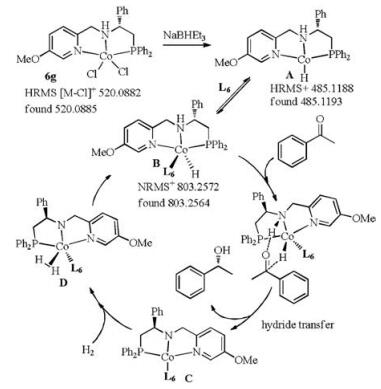
In order to gain some understanding of the catalytic mechanism and particularly the role of the phosphine additive, electrospray ionization high-resolution mass spectrometry (ESI-HRMS) was employed to determine possible intermediates. The results are summarized in Fig. S2 (Supporting information). Firstly, a high-intensity signal of the Co(Ⅱ) complex 6g at the m/z value of 520.0885 corresponding to [M-Cl]+ was observed (Fig. S2). With the addition of two equivalents of NaBHEt3, a high-intensity signal of an Co(I)-H hydride intermediate A at the m/z value of 485.1193 for [M-hydride]+ was observed (Fig. S2). Following this, three equivalents of phosphine L6 was added. As can be seen from the mass spectra, one signal at the m/z value of 803.2564 assigned to [M+L6+H]+ was observed. This might be due to the coordination of L6 to the Co-hydride to form B. Considering these HRMS results, the asymmetric hydrogenation reaction of ketones may proceed as follows (Scheme 4).

|
Download:
|
| Scheme 4. Proposal mechanism. | |
{kind=link}
The Co(Ⅱ) complex 6g is reduced to Co(I) with NaBHEt3 [5h], affording a 16e− Co-hydride species A. The phosphine L6 ligand coordinates to A giving an 18e− Co(I)-hydride-L6 species B. With the addition of a substrate ketone, the reduction proceeds via an outer-sphere proton-hydride transfer mechanism as in Noyori's DEPN-Ru system, involving the assistance of the NH functional group on the ligand to hydrogen-bond with the substrate [10]. The chiral alcohol is produced along with the 16e− species C. The dihydrogen complex D can be generated from C under H2. Finally, the active species B is regenerated via dihydrogen activation under the assistance of the amido nitrogen.
In conclusion, we have found a PNN-Co complex to catalyze the hydrogenation of ketones. Upon addition of a free mono-phosphine ligand, the complex turns into highly efficient enantioselective catalyst, allowing a broad scope of ketones to be reduced to chiral alcohols with up to 93% enantioselectivity. Whilst the role of the mono-phosphine remains to be elucidated, its remarkable effect in this PNN-Co catalytic system may afford a useful clue to the design of more effective chiral cobalt catalysts in the future.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentWe are grateful for the financial support of the National Natural Science Foundation of China (No. 21672133).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.011.
| [1] |
(a) C.S.G. Seo, R.H. Morris, Organometallics 38 (2019) 47-65; (b) J.D. Hayler, D.K. Leahy, E.M. Simmons, Organometallics 38 (2019) 36-46; (c) J. Magano, J.R. Dunetz, Org. Process Res. Dev. 16 (2012) 1156-1184; (d) F.D. Klingler, Acc. Chem. Res. 40 (2007) 1367-1376; (e) W.S. Knowles, Angew. Chem., Int. Ed. 41 (2002) 1998-2007. |
| [2] |
(a) W. Ai, R. Zhong, X. Liu, et al., Chem. Rev. 119 (2019) 2876-2953; (b) W. Liu, B. Sahoo, K. Junge, et al., Acc. Chem. Res. 51 (2018) 1858-1869; (c) G.A. Filonenko, R. van Putten, E.J.M. Hensen, et al., Chem. Soc. Rev. 47 (2018) 1459-1483; (d) B. Maji, M.K. Barman, Synthesis 49 (2017) 3377-3393; (e) J.L. Renaud, S. Gaillard, Synthesis 48 (2016) 3659-3683; (f) Y.Y. Li, S.L. Yu, W.Y. Shen, et al., Acc. Chem. Res. 48 (2015) 2587-2598; (g) H. Pellissier, H. Clavier, Chem. Rev. 114 (2014) 2775-2823. |
| [3] |
(a) R. Huber, A. Passera, E. Gubler, et al., Adv. Synth. Catal. 360 (2018) 2900-2913; (b) S.A.M. Smith, P.O. Lagaditis, A. Luepke, et al., Chem. Eur. J. 23 (2017) 7212-7216; (c) P.O. Lagaditis, P.E. Sues, J.F. Sonnenberg, et al., J. Am. Chem. Soc. 136 (2014) 1367-1380; (d) Y. Li, S. Yu, X. Wu, et al., J. Am. Chem. Soc. 136 (2014) 4031-4039; (e) Y. Hu, Z. Zhang, J. Zhang, et al., Angew. Chem. Int. Ed. 58 (2019) 15767-15771; (f) D. Zhang, E.Z. Zhu, Z.W. Lin, et al., Asian J. Org. Chem. 5 (2016) 1323-1326; (g) G. Liu, X. Zhang, H. Wang, et al., Chem. Commun. 56 (2020) 4934-4937; (h) B. Li, J. Chen, Z. Zhang, et al., Angew. Chem. Int. Ed. 58 (2019) 7329-7334; (i) Y. Liu, Z. Yi, X. Tan, et al., iScience 19 (2019) 63-73; (j) W. Gao, H. Lv, T. Zhang, et al., Chem. Sci. 8 (2017) 6419-6422; (k) L. Zhang, Y. Tang, Z. Han, et al., Angew. Chem. Int. Ed. 58 (2019) 4973-4977; (l) F. Ling, H. Hou, J. Chen, et al., Org. Lett. 21 (2019) 3937-3941. |
| [4] |
(a) F.K. Shmidt, Y.S. Levkovskii, N.M. Ryutina, et al., React. Kinet. Catal. Lett. 12 (1979) 475-478; (b) L. Simandi, E. Budo, Acta Chim. 64 (1970) 125-138. |
| [5] |
(a) H. Zhong, M.R. Friedfeld, P.J. Chirik, Angew. Chem. Int. Ed. 58 (2019) 9194-9198; (b) M.R. Friedfeld, H. Zhong, R.T. Ruck, et al., Science 360 (2018) 888-889; (c) J. Guo, X. Shen, Z. Lu, Angew. Chem., Int. Ed. 56 (2017) 615-618; (d) J. Guo, B. Cheng, X. Shen, et al., J. Am. Chem. Soc. 139 (2017) 15316-15319; (e) M.R. Friedfeld, M. Shevlin, G.W. Margulieux, et al., J. Am. Chem. Soc. 138 (2016) 3314-3324; (f) J. Chen, C. Chen, C. Ji, et al., Org. Lett. 18 (2016) 1594-1597; (g) M.R. Friedfeld, M. Shevlin, J.M. Hoyt, et al., Science 342 (2013) 1076-1080; (h) S. Monfette, Z.R. Turner, S.P. Semproni, et al., J. Am. Chem. Soc. 134 (2012) 4561-4564. |
| [6] |
(a) R. Zhong, Z. Wei, W. Zhang, et al., Chem 5 (2019) 1552-1566; (b) Z. Shao, R. Zhong, R. Ferraccioli, et al., Chin. J. Chem. 37 (2019) 1125-1130; (c) S. Sandl, F. Schwarzhuber, S. Poellath, et al., Chem. Eur. J. 24 (2018) 3403-3407; (d) S. Roesler, J. Obenauf, R. Kempe, J. Am. Chem. Soc. 137 (2015) 7998-8001; (e) D. Gaertner, A. Welther, B.R. Rad, et al., Angew. Chem. Int. Ed. 53 (2014) 3722-3726; (f) G. Zhang, K.V. Vasudevan, B.L. Scott, et al., J. Am. Chem. Soc. 135 (2013) 8668-8681; (g) T.P. Lin, J.C. Peters, J. Am. Chem. Soc. 135 (2013) 15310-15313; (h) G. Zhang, B.L. Scott, S.K. Hanson, Angew. Chem. Int. Ed. 51 (2012) 12102-12106; (i) V. Massonneau, P. Le Maux, G. Simonneaux, J. Organomet. Chem. 288 (1985) C59-C60. |
| [7] |
(a) R. Adam, J.R. Cabrero-Antonino, A. Spannenberg, et al., Angew. Chem. Int. Ed. 56 (2017) 3216-3220; (b) R. Adam, C.B. Bheeter, J.R. Cabrero-Antonino, et al., ChemSusChem 10 (2017) 842-846. |
| [8] |
(a) Z. Shao, S. Fu, M. Wei, et al., Angew. Chem. Int. Ed. 55 (2016) 14653-14657; (b) S. Fu, N.Y. Chen, X. Liu, et al., J. Am. Chem. Soc. 138 (2016) 8588-8594; (c) M.V. Gradiski, B.T.H. Tsui, A.J. Lough, et al., Dalton Trans. 48 (2019) 2150-2159. |
| [9] |
(a) R. Rexiti, Z.G. Zhang, J. Lu, et al., J. Org. Chem. 84 (2019) 1330-1338; (b) B. Pan, B. Liu, E. Yue, et al., ACS Catal. 6 (2016) 1247-1253; (c) T. Morimoto, Y. Yamaguchi, M. Suzuki, et al., Tetrahedron Lett. 41 (2000) 10025-10029. |
| [10] |
T. Ohkuma, N. Utsumi, K. Tsutsumi, et al., J. Am. Chem. Soc. 128 (2006) 8724-8725. |