 2021, Vol. 32
2021, Vol. 32 


Oxocarbenium ions are key intermediates in a variety of synthetic transformations and in the chemistry of carbohydrates in particular [1]. Catalytic asymmetric addition of carbon nucleophiles to prochiral oxocarbenium ions has remained challenging because they typically lack a Lewis basic site for interaction with chiral catalyst [2]. During the past decade, significant advances have been achieved in enantioselective intermolecular additions to cyclic oxocarbenium ions [3]. In sharp contrast, catalytic asymmetric intermolecular addition to acyclic species has remained undeveloped, presumably due to the lack of rigid geometry [4]. In 2008, Sodeoka disclosed a Pd(Ⅱ)-catalyzed asymmetric addition of β-ketoesters to acyclic, alkenyl-stabilized oxocarbenium ions generated in situ via collapse of acetals [4a]. In 2011, Kobayashi reported a chiral Nb-catalyzed enantioselective addition of silyl enol ethers to acyclic oxocarbenium ions [4b]. The groups of Lambert and Jacobsen respectively documented delicate organo-catalyzed asymmetric alkylation of acyclic, aryl-stabilized oxocarbenium ions with corresponding silyl enol ethers [4c, 4d]. Despite great innovation and high enantiocontrol, the specific structures of above mentioned nucleophiles (silyl enol ethers and β-ketoesters) restrict the integrated pattern of functionalities in the α-position of ether derivatives.
Enantiopure β-alkoxyl carbonyl moieties with diverse α-substituents are common building blocks in a number of natural products. N-Acetyl thiazolidinethiones derived from diverse carboxylic acids have been widely used as versatile nucleophiles for aldol reactions of aldehydes to prepare such building blocks through a two-step protocol in complex molecule synthesis [5].
Moreover, since the pioneering work of Urpí, a one-step asymmetric aldol-type addition of chiral N-acetyl thiazolidinethiones onto acetals for direct access to chiral β-alkoxyl carbonyls with diverse α-alkyl moieties has been explored [6]. However, catalytic asymmetric variants have been less investigated [7]. Therefore, the development of a catalytic asymmetric alkylation of acyclic oxocarbenium ions generated in situ from acetal precursors with diverse carboxylic acid derivatives would enrich the chemistry of oxocarbenium ions and provide an economic and effective method to prepare enantiopure β-alkoxyl carbonyls with diverse α-substituents.
The asymmetric alkylation of benzaldehyde dimethyl acetal 1a with N-(phenylacetyl)oxazolidine-2-thione 2a was initially selected as a model reaction for optimization using bisoxazoline L1 (Fig. 1) as chiral ligand, BF3·OEt2 as acidic additive, and 2, 6-lutidine as basic additive in CH2Cl2 (Table 1). A variety of metal salt catalysts were evaluated, and Ni(OTf)2 was identified to be optimal (Table 1, entries 1–4). A further screen of chiral ligands suggested that BINAP L3 (Fig. 1) furnished the best yields and stereoselectivities (entries 3 and 5–8). Other nickel(Ⅱ) catalysts such as Ni(OAc)2 and NiBr2 together with copper(Ⅱ) catalysts such as Cu(OTf)2 afforded inferior results to Ni(OTf)2 (entries 6 and 9–11). The combination of BF3·OEt2/2, 6-lutidine proved to be crucial to both the reactivity and ee, and replacing either component with TMSOTf or Et3N was sterile (entries 12–14). The alkylation using pre-prepared L3/Ni(OTf)2 complex provided a significant improvement in reaction yield (entries 6 and 15). The ratio of BF3·OEt2/2, 6-lutidine was also found to be very important, and the best results were observed for reaction with excess of acidic additive (entries 15–18). Solvent optimization identified a mixed solvents of CH2Cl2/THF (v: v=1:3) to be optimal, and the expected3a/3a' were isolated in 83% yield and 98% ee/97% ee as a separable mixture of diastereomers (d.r.=1.8:1) (entry 19).

|
Download:
|
| Fig. 1. The chiral ligands. | |
|
|
Table 1 Reaction condition optimization.a |
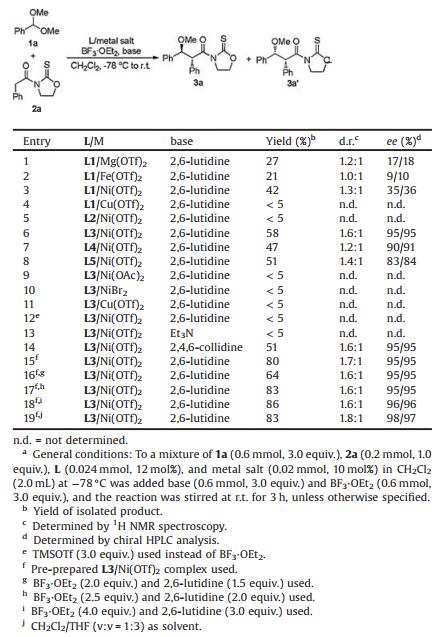
{kind=link}
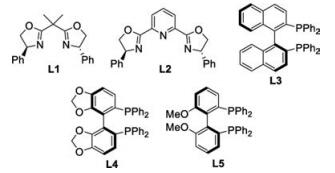
With the optimized reaction conditions in hand, the scope of carboxylic acid derivatives was investigated (Scheme 1). While N-(phenylacetyl)-2-oxazolidinone 2c proved to be an invalid nucleophilic component, N-(phenylacetyl)oxazolidine-thione 2a and N-(phenylacetyl)-thiazolidine-thione 2b were well compatible with the alkylation conditions, with the former to be optimal in terms of efficiency and stereoselectivities (Scheme 1A). A broad range of N-(acetyl)oxazolidine-thiones 2d-2i bearing electronically varied aryl groups with diverse substituent patterns were tolerated, furnishing corresponding 3d-3i in good yields (75%–83%) with excellent ee (95%–97%) and moderate diastereocontrol. α-Alkenyl-substituted acetic acid derivatives were suitable coupling partners (Scheme 1B). γ-Aryl substituted 3-butenoyl 2j and 2k together with γ-alkyl substituted 2l and 2m were competent substrates, and respective 3j-3m were obtained in good yields with 91%–94% ee. Notably, all the reactions showed excellent regioselectivity, and the alkylation occurred exclusively at the α-position of 3-butenoyl derivatives with olefin geometry highly conserved. N-(Alkylacetyl)oxazolidine-thiones 2n and 2p together with N-(alkylacetyl)-thiazolidine-thione 2o were also well tolerated (Scheme 1C).

|
Download:
|
| Scheme 1. The scope of carboxylic acid derivatives. d.r. was calculated by separating and weighing two diastereomers. | |
{kind=link}
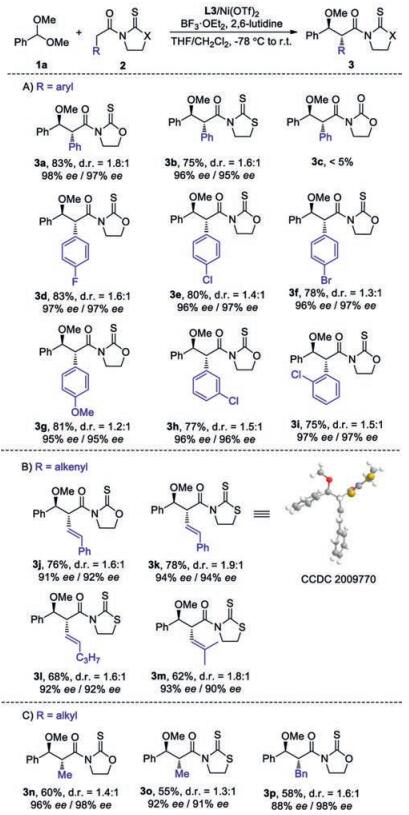
The scope of acetal moieties was then explored (Scheme 2). A variety of aryl-aldehyde dimethyl acetals 1 bearing electronically varied aryl groups with diverse substituent patterns were well tolerated, furnishing corresponding 4a-4g in good yields (73%–91%) with excellent ee (92%–98%) and moderate diastereocontrol (Scheme 2A). Heteroaryl aldehyde derived acetal was also suitable substrate, as demonstrated by the generation of 4h in 75% yield with 92% ee. Dimethyl acetals derived from β-aryl and -alkyl substituted acrylaldehydes were also competent substrates, as illustrated by the isolation of 4i-4n in high efficiency with excellent ee (Scheme 2B).

|
Download:
|
| Scheme 2. The scope of acetal components. d.r. was calculated by separating and weighing two diastereomers. | |
{kind=link}
A gram-scale asymmetric alkylation of acetal 1a with 2a was conducted without obvious loss of ee as well as efficiency, as illustrated by the generation of 3a in 75% yield with 96% ee/95% ee (d.r.=1.6:1) (Scheme 3A). The oxazolidinethione 3a' can be facilely transformed to synthetically valuable methyl ester 5 and weinreb amide 6, respectively (Schemes 3B and C). Oxazolidinethione can also be reduced by LiBH4 furnishing primary alcohol 7 (Scheme 3D).

|
Download:
|
| Scheme 3. Synthetic utilities. | |
{kind=link}
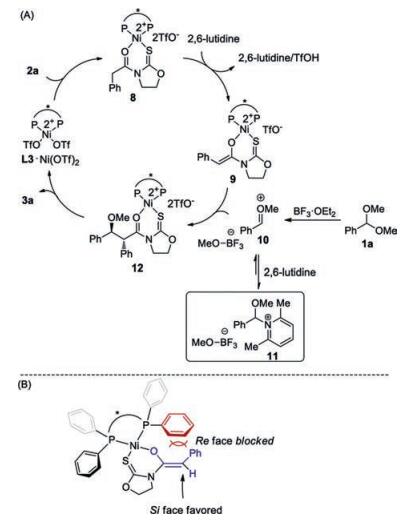
The mechanism for nickel(Ⅱ)-catalyzed asymmetric alkylation of benzaldehyde dimethyl acetal 1a with N-(phenylacetyl)oxazolidine-2-thione 2a was proposed (Scheme 4A). 2a coordinates with prepared L3/Ni(OTf)2 catalyst giving 8. In complex 8, the hydrogen α to the carbonyl moiety was activated in terms of the acidity. 2, 6-Lutidine facilitates the enolization of 8 furnishing chiral Ni-bound Z-enolate 9 [8].9 attacks acyclic oxocarbenium 10 generated in situ from 1a and BF3·OEt2 providing 12. Product dissociation completes the catalytic cycle. No reactivity was observed when Et3N was used instead of 2, 6-lutidine or 2, 4, 6-collidine, suggesting that the role of pyridine derivatives might not simply act as a base (entries 6, 13 and 14, Table 1) [9]. It has been known that acetal might react with 2, 4, 6-collidine affording corresponding pyridinium-type salt [10, 7b]. Accordingly, we envisioned that 2, 6-lutidine might react with acyclic oxocarbenium10 reversibly giving pyridinium 11. 11 possessing weak electrophilicity might be regarded as a reservoir of 10 to prevent the decomposition before the attack by enolate 9. The stereochemical induction model was proposed in Scheme 4B [7]. The P-bound phenyl group on the ligand (in red) shields the top face (Re face) of the enolate substrate (in blue), which disfavors the electrophilic attack by the oxocarbenium intermediate. In contrast, the addition of the oxocarbenium ion to the Si face of the enolate substrate is favored because of less repulsive interactions with the ligand. The observed moderate diastereoselectivity might be ascribed to the lack of Lewis basic site on corresponding oxocarbenium ion intermediates for substrate–catalyst interactions.

|
Download:
|
| Scheme 4. Proposed reaction mechanism and stereochemistry analysis. | |
{kind=link}
Based on above analysis, we envisioned that placing a germinal substitution at the C4 of 1, 3-oxazolidine-2-thione might be beneficial for improving the d.r.. Under the standard conditions, asymmetric alkylation of acetal 1a with 13 bearing a germinal dimethyl substitution proceeded, furnishing expected 14 in 78% yield and 98% ee/97% ee as a separable mixture of diastereomers (d.r.=2.6:1), indicating that modification of 1, 3-oxazolidine-2-thione might be a viable approach for enhancing the diastereoselectivity (Scheme 5).

|
Download:
|
| Scheme 5. Substituent effect on 1, 3-oxazolidine-2-thione. | |
{kind=link}
In conclusion, a nickel(Ⅱ)-catalyzed asymmetric alkylation of acyclic oxocarbenium ions generated in situ from corresponding acetals with carboxylic acid derivatives to prepare β-alkoxyl carbonyl moieties with diverse α-substituents has been disclosed. The method exhibited broad scope of acetals and carboxylic acid derivatives with excellent enantioselectivity and good functional group compatibility, and can be conducted in a gram-scale without obvious loss of efficiency.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financial supported by the Natural Science Foundation of China (Nos. 21722204, 21971148) and Fok Ying Tung Education Foundation (No. 151035).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.08.034.
| [1] |
L. Krasnova, C.H. Wong, Annu. Rev. Biochem. 85 (2016) 599-630. DOI:10.1146/annurev-biochem-060614-034420 |
| [2] |
(a) L. Liu, P.S.J. Kaib, A. Tap, B. List, J. Am. Chem. Soc. 138 (2016) 10822–10825; (b) Y. Xie, G.J. Cheng, S. Lee, et al., J. Am. Chem. Soc. 138 (2016) 14538–14541; (c) C. Zhao, S.B. Chen, D. Seidel, J. Am. Chem. Soc. 138 (2016) 9053–9056; (d) K. Kanomata, Y. Toda, Y. Shibata, et al., Chem. Sci. 5 (2014) 3515–3523; (e) C.D. Gheewala, J.S. Hirschi, W.H. Lee, et al., J. Am. Chem. Soc. 140 (2018) 3523–3527; (f) A. Lee, R.C. Betori, E.A. Crane, K.A. Scheidt, J. Am. Chem. Soc. 140 (2018) 6212–6216; (g) R. Zhao, G. Feng, X. Xin, et al., Chin. Chem. Lett. 30 (2019) 1432–1434; (h) Z. Wang, Y. Mao, H. Guan, et al., Chin. Chem. Lett. 30 (2019) 1241–1243. |
| [3] |
(a) S.E. Reisman, A.G. Doyle, E.N. Jacobsen, J. Am. Chem. Soc. 130 (2008) 7198-7199; (b) S. Lee, P.S.J. Kaib, B. List, J. Am. Chem. Soc. 139 (2017) 2156–2159; (c) P.N. Moquist, T. Kodama, S.E. Schaus, Angew. Chem. Int. Ed. 49 (2010) 7096-7100; (d) M.H.D. Srinivas, M.P. Watson, J. Am. Chem. Soc. 133 (2011) 17142–17145; (e) F. Benfatti, E. Benedetto, P.G. Cozzi, Chem. Asian J. 5 (2010) 2047–2052; (f) M. Rueping, C.M.R. Volla, I. Atodiresei, Org. Lett. 14 (2012) 4642–4645; (g) S. Dasgupta, T. Rivas, M.P. Watson, Angew. Chem. Int. Ed. 54 (2015) 14154-14158; (h) Z.Y. Han, R. Guo, P.S. Wang, D.F. Chen, H. Xiao, L.Z. Gong, Tetrahedron Lett. 52 (2011) 5963–5967; (i) Y. Cui, L.A. Villafane, D.J. Clausen, P.E. Floreancig, Tetrahedron 69 (2013) 7618–7626; (j) Z. Meng, S. Sun, H. Yuan, H. Lou, L. Liu, Angew. Chem. Int. Ed. 53 (2014) 543-547; (k) X. Pan, X. Liu, S. Sun, Z. Meng, L. Liu, Chin. J. Chem. 36 (2018) 1187–1190; (l) X. Xin, X. Pan, Z. Meng, X. Liu, L. Liu, Org. Chem. Front. 6 (2019) 1448–1452; (m) H. Guan, L. Chen, L. Liu, Acta Chim. Sinica 76 (2018) 440–444; (n) A. Gualandi, G. Rodeghiero, P.G. Cozzi, Asian J. Org. Chem. 7 (2018) 1957-1981; (o) M. Braun, W. Kotter, Angew. Chem. Int. Ed. 43 (2014) 514–517; (p) L. Wang, D. Yang, D. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 9088–9092. |
| [4] |
(a) N. Umebayashi, Y. Hamashima, D. Hashizume, M. Sodeoka, Angew. Chem. Int. Ed. 47 (2008) 4196–4199; (b) S. Kobayashi, K. Arai, T. Yamakawa, et al., Adv. Synth. Catal. 353 (2011) 1927–1932; (c) C.D. Gheewala, B.E. Collins, T.H. Lambert, Science 351 (2016) 961–965; (d) S.M. Banik, A. Lvvina, A.M. Hyde, E.N. Jacobsen, Science 358 (2017) 761–764; (e) C. Lu, X. Su, P.E. Floreancig, J. Org. Chem. 78 (2013) 9366–9367. |
| [5] |
(a) D.A. Evans, J. Bartroli, T.L. Shih, J. Am. Chem. Soc. 103 (1981) 2127–2129; (b) D.A. Evans, J.M. Takacs, L.R. McGee, et al., Pure Appl. Chem. 53 (1981) 1109-1127; (c) M.M. Heravi, V. Zadsirjan, Tetrahedron Asymmetry 24 (2013) 1149–1188. |
| [6] |
(a) A. Cosp, P. Romea, P. Talavera, et al., Org. Lett. 3 (2001) 615–617; (b) T.E. Smith, W.H. Kuo, E.P. Balskus, et al., J. Org. Chem. 73 (2008) 142–150; (c) T.E. Smith, W.H. Kuo, V.D. Bock, et al., Org. Lett. 9 (2007) 1153–1155; (d) K.K. Pulukuri, T.K. Chakraborty, Org. Lett. 14 (2012) 2858–2861; (e) M.T. Crimmins, C.O. Hughes, Org. Lett. 14 (2012) 2168–2171; (f) J. Fernández-Valparis, P. Romea, F. Urpí, M. Font-Bardia, Org. Lett. 19 (2017) 6400–6403; (g) J.M. Romo, E. Gálvez, I. Nubiola, et al., Adv. Synth. Catal. 355 (2013) 2781-2786; (h) S.C.D. Kennington, J.M. Romo, P. Romea, F. Urpí, Org. Lett. 18 (2016) 3018-3021; (i) T.K. Kuilya, R.K. Goswami, Org. Lett. 19 (2017) 2366–2369. |
| [7] |
(a) D.A. Evans, R.J. Thomson, J. Am. Chem. Soc. 127 (2005) 10506–10507; (b) G. Wang, X. Xin, Z. Wang, et al., Nature Commun. 10 (2019) 559–567. |
| [8] |
D.A. Evans, C.W. Downey, J.T. Shaw, J.S. Tedrow, Org. Lett. 4 (2002) 1127-1130. DOI:10.1021/ol025553o |
| [9] |
(a) F. Brotzel, B. Kempf, T. Singer, H. Zipse, H. Mayr, Chem. Eur. J. 13 (2007) 336-345; (b) J. Ammer, M. Baidya, S. Kobayashi, H. Mayr, J. Phys. Org. Chem. 23 (2010) 1029–1035. |
| [10] |
H. Fujioka, T. Okitsu, Y. Sawama, et al., J. Am. Chem. Soc. 128 (2006) 5930-5938. DOI:10.1021/ja060328d |