 2021, Vol. 32
2021, Vol. 32 


b Zhejiang Provincial Engineering Research Center of Industrial Boiler & Furnace Flue Gas Pollution Control, Hangzhou 310058, China;
c Department of Environmental Engineering, China Jiliang University, Hangzhou 310018, China
Large quantities of chlorinated volatile organic compounds (CVOCs) are frequently emitted into the environment from many industrial processes [1, 2] which serve as irreplaceable solvents, cleaning agents, and intermediates in the production such as pharmaceutical intermediates [3-5]. CVOCs such as dichloromethane (DCM), 1, 2-dichloroethane (DCE), trichloroethylene (TCE), and chlorobenzene (CB) [6, 7] do harm to both human health [8] and the environment [9, 10]. Among the various methods for CVOCs removal, catalytic oxidation is considered as an economical and effective technology [11, 12].
At present, significant efforts are made to investigate noble metal catalysts, transition metal oxide catalysts and zeolite catalysts for catalytic oxidation of CVOCs. Noble metal catalysts are high active and exhibit good selectivity for COx during the reaction. But their high price and quick deactivation by chlorine poisoning still remain disadvantages [13]. Transition metal oxides have gained lots of considerations because of their low price, noticeable resistance to chlorine poisoning, and good oxidative activities [14]. Particularly, Co3O4 has attracted considerable interest due to its high activity in the total oxidation of VOCs [15]. Co3O4 has a typical spinel structure comprising Co2+ at the tetrahedral sites and Co3+ at the octahedral sites [16]. Cobalt oxides have a high bulk oxygen mobility and can easily form highly active oxygen species (O− or O2−) which make them active catalysts [17]. However, Co3O4 has some disadvantages such as structural instability at high temperatures [18] as well as easy chlorination when during CVOC oxidation [16, 19]. Zeolites have good characteristics such as a high surface area, acidic properties, unique pore structures, and good thermal stability [20, 21]. Nevertheless, with the reaction time increases, acid zeolites are deactivated because of coke deposition and chlorine poisoning for CVOC elimination [22].
Studies have reported that a good strategy is to combine transition metal oxides and zeolites to prepare a catalyst. Divakar et al. [23] developed several Fe-zeolite catalysts for the trichloroethylene (TCE) elimination. The test results showed that the activity of zeolites was enhanced because of the addition of Fe. The fabrication procedure of the catalyst also affects the performance of catalysts. Samples prepared by impregnation and solid state ion exchange methods are more active than the samples prepared just by the impregnation method. Blanch-Ragaet al. [24] developed Cu and Co beta zeolite catalysts and found that Cu-exchanged zeolite beta catalysts had the highest activity for the oxidation of TCE. The preparation method significantly affects catalytic properties. Therefore, it is envisaged that cobalt oxides and zeolites can be reasonably combined by certain preparation methods for the elimination of CVOCs.
The synthesis method of catalysts affects its crystals size and other properties [25]. The hydrothermal method is an important technique to prepare nanomaterials and has been widely researched during recent years [26, 27]. With the wide application of the hydrothermal method, techniques such as microwaves, mechanical mixing, and electric fields are used to achieve the target product [28]. The microwave hydrothermal method is used to prepared samples under pressurized conditions in a closed reaction system. In comparison with the conventional hydrothermal synthesis, microwave irradiation can produce efficient internal heating which dramatically reduces reaction time [29]. Hence, the microwave hydrothermal synthesis has the characteristics of a rapid heating speed, sensitive reaction, and uniform heating system, which lead to the rapid synthesis nanoparticles with uniform morphology and narrow particle size distribution [28]. The reactants in conventional hydrothermal synthesis usually lack frequent contact because they are precipitated at the bottom of the reactor [30]. Dynamic hydrothermal methods can overcome this limitation of static hydrothermal synthetic methods [31].
In this work, three Co3O4-ZSM-5 catalysts named CH-Co3O4/ZSM-5 (CH for short), MH-Co3O4@ZSM-5 (MH for short), and DH-Co3O4@ZSM-5 (DH for short) were prepare by the conventional hydrothermal method, microwave hydrothermal method, and dynamic hydrothermal method and their catalytic oxidation of dichloromethane was investigated. The physicochemical properties of different samples were characterized by X-ray diffractometer (XRD), scanning microscopy (SEM), X-ray photoelectron spectroscopy (XPS), Brunauer–Emmett–Teller (BET), H2 temperature-programmed reduction (H2-TPR), temperature-programmed desorption of O2 (O2-TPD), temperature-programmed desorption of NH3 (NH3-TPD), diffuse reflectance infrared Fourier-transform spectra with NH3 molecules (NH3-DRIFT), and temperature-programmed surface reaction (TPSR) to identify the impact of different synthetic methods on the catalysts. Based on the in situ DRIFT tests, a plausible DCM oxidation mechanism was proposed over the MH-Co3O4@ZSM-5 catalysts.
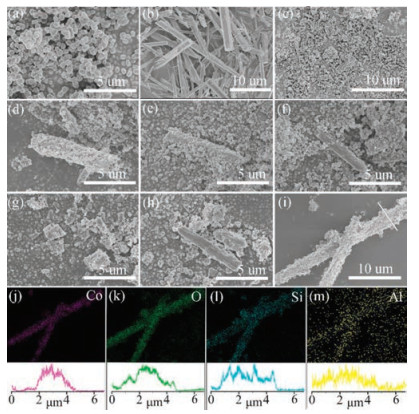
Morphologies of all the samples were investigated with SEM (Fig. 1). ZSM-5 obtained an ovoid or ellipsoid-shaped structure (about 200nm long and 400nm wide) with rough surface. Co3O4 nanorods (Fig. 1b) were calcined into debris (Fig. 1c) because of the high temperature. Using the dynamic hydrothermal method, we found that some of the Co3O4 nanorods were totally wrapped by ZSM-5 nanoparticles and formed the core-shell structure (Fig. 1d), some were partly wrapped by ZSM-5 (Fig. 1e), and some were almost bare and broken (Fig. 1f). The dynamic hydrothermal method could not synthesize samples of uniform morphology. For the samples prepared by conventional hydrothermal synthesis, the Co3O4 nanorods were calcined into debris, while ZSM-5 nanoparticles could not form around the surface of Co3O4 (Figs. 1g and h). Fig. 1i shows that the microwave hydrothermal method achieved uniform distribution of a monolayer of spherical ZSM-5 nanoparticles on the Co3O4 nanorod surface and formed a core-shell structure. With the protection of the ZSM-5 shell, Co3O4 nanorods maintained their original appearance. The energy-dispersive X-ray spectroscopy (EDS) elemental mapping of MH-Co3O4@ZSM-5 confirmed the uniform distribution of O, Si, and Al across the cobalt oxide nanorods without segregation (Figs. 1j–m). EDS line-scan profiles showed the core-shell of the sample.

|
Download:
|
| Fig. 1. SEM images of (a) ZSM-5, (b) Co3O4 before calcination, (c) Co3O4 after calcination, (d-f) DH-Co3O4@ZSM-5, (g, h) CH-Co3O4/ZSM-5 and (i) MH-Co3O4@ZSM-5. (j-m) EDS-mapping images of different elements and EDS line-scan profiles of MH-Co3O4@ZSM-5. | |
{kind=link}
Fig. S1 (Supporting information) depicts the XRD patterns of the tested samples. The diffraction peaks of Co3O4 sample at 2θ of 19.0°, 31.3°, 36.8°, 38.5°, 44.8°, 59.4° and 65.2° were corresponding to the phase of spinel Co3O4 phase (JCPDS PDF # 42-1467) [32]. The peaks at 7°–9° and 23°–25° were indexed to the ZSM-5 (JCPDS PDF # 44-0003) [33]. The Co3O4 and ZSM-5 phases were both observed in the three Co3O4-ZSM-5 samples and no peaks ascribed to other phases or impurities were observed. In addition, the average crystal sizes of the Co3O4-ZSM-5 samples were calculated according to the Scherrer equation. The crystal sizes of MT, DT, and CT samples were 49.2nm, 33.9nm and 32.2nm, respectively. The difference of crystal sizes could be attributed to the various preparation conditions. The CT sample obtained the smallest crystal size which was consistent with the SEM figure because the Co3O4 was fractured into debris after the calcination. MT-Co3O4@ZSM-5 showed the largest crystal size because of the protection of the ZSM-5 shell which maintained the rod shape of the Co3O4.
Different fabrication methods lead to differences in textural properties of the catalysts (Table S1 in Supporting information). Specific areas of ZSM-5 and three Co3O4-ZSM-5 samples were much higher than those of Co3O4 (6.8m2/g). Furthermore, MH (257.3m2/g) and DH (257.6m2/g) samples showed the higher surface areas than CH-Co3O4/ZSM-5 (239.0m 2/g) because the reactants of the conventional hydrothermal method were lacking in frequent contact and sank at the bottom of the reactor, leading to lower ZSM-5 yield. For the dynamic hydrothermal synthesis, a ZSM-5 shell was formed by a reaction between the surface hydroxy groups of Co3O4 with highly reactive and hydrolyzed tetraethyl orthosilicate (TEOS) [34]. In microwave hydrothermal process, Co3O4 nanorods could absorb microwaves and convert the energy into heat, which led to the generation of surface "super hot" dots [35]. The "super hot" dots could facilitate the adsorption of more hydrolyzed TEOS, leading to the higher yield of ZSM-5. Therefore, it was speculated that different synthetic methods lead to the variation of ZSM-5 content in the Co3O4-ZSM-5 and result in the different surface areas.
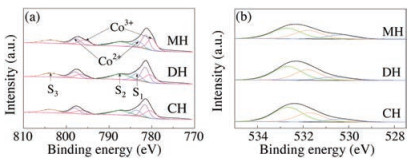
XPS analysis was carried out to investigate the elemental chemical states and surface oxygen species of the samples. Fig. 2 depicts the Co 2p and O 1s XPS spectra of the catalysts. Fig. 2a presents the spectra of Co 2p, in which two main signals of Co 2p3/2 and Co 2p1/2 were observed at around 781.2eV and 796.9eV, respectively. The Co 2p spectra of the three catalysts could be deconvoluted into two spin-orbit doublets (Co3+ (D1) and Co2+ (D2)) and three satellite peaks (S1, S2, and S3) [36]. The deconvolution of Co 2p3/2 showed peaks at 782.0±0.2eV and 781.5±0.1eV, which could be respectively ascribed to Co2+ and Co3+ cations [37]. Moreover, the fitted ratio of Co3+/Co2+ in MH-Co3O4@ZSM-5 (1.00) was higher than those in DH-Co3O4@ZSM-5 (0.78) and CH-Co3O4/ZSM-5 (0.81), indicating that abundant Co3+ species existed in the samples. The higher number of Co3+ species might have increased the anionic defects, which contributed to the absorption and activation of oxygen in the gas phase, corresponding to the higher catalytic activity [38].

|
Download:
|
| Fig. 2. XPS patterns of the catalysts, (a) Co 2p and (b) O 1s. | |
{kind=link}
The O 1s spectra of the three catalysts (Fig. 2b) could be deconvoluted into three peaks at about 530.5, 531.8, and 532.7eV, which were ascribed to surface lattice oxygen species (Olatt), surface adsorbed oxygen species (Oads), and chemisorbed water (Ow), respectively [39, 40]. The adsorbed oxygen species were helpful for activation of oxygen molecules and were more conducive to the deep oxidation of CVOCs [41, 42]. Therefore, Oads/(Oads + Olatt + Ow) is showed in Table S2 (Supporting information) according to the peak areas. As presented in Table S2, the percentage of Oads over all the samples was basically the same. For the MH sample, the percentage of Oads was slightly higher than that of the other two catalysts, suggesting that MH-Co3O4@ZSM-5 had more surface adsorbed oxygen species.
XPS studies also show the content of cobalt (7.9%−10.3%), aluminum (0.5%−1.7%), and silicon (21.6%−23.9%) in the three samples. The MH samples obtained the lowest Co content and the highest Al/Si content among the three catalysts. It indicated that there were less Co and more Al/Si on the surface of MH-Co3O4@ZSM-5, corresponding to its core-shell structure.
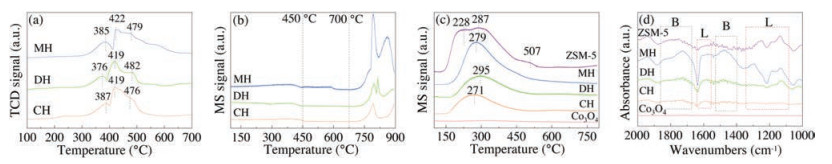
As depicted in Fig. 3a, H2-TPR was carried out to investigate the redox properties of the samples. For the MH-Co3O4@ZSM-5 catalyst, the reduction peak centered at around 385 ℃ assigned to the reduction process from Co3+ to Co2+ (Co3O4 + H2 → 3CoO + H2O), while the peaks centered at 422 ℃ and 479 ℃ was attributed to the reduction of Co2+ to Co0 (3CoO + 3H2 → 3Co + 3H2O) [38]. Generally, the hydrogen consumption is of great significance for the sample's catalytic performance [14]. It could be observed that the intensity for the first peak at around 385 ℃ of the MH sample was greater than that of the other two samples. This showed that MH-Co3O4@ZSM-5 possessed higher quantities of Co3+ [43], which was also conformed from the XPS test above. The intensities for the second peak of the three samples centered around 420 ℃ seemed considerably close, while the third peak centered at approximately 479 ℃ of the MH catalyst was the strongest. The observations indicated that the sample prepared by the microwave hydrothermal method exhibited the best low temperature reducibility than the samples fabricated by the other two methods, which was closely related to the better catalytic activity [44].

|
Download:
|
| Fig. 3. (a) H2-TPR, (b) O2-TPD, (c) NH3-TPD, and (d) NH3-DRIFT profiles of the catalyst. | |
{kind=link}
O2-TPD was used to investigate the interaction between adsorbed oxygen species and the Co3O4-ZSM-5 surface (Fig. 3b). The desorption peaks below 450 ℃, between 450–550 ℃, and above 550 ℃ could be ascribed to the evolution of surface adsorbed oxygen species, surface lattice oxygen species, and bulk lattice oxygen species, respectively [45, 46]. It was reported that larger desorption peak area of surface reactive oxygen species at low temperatures indicated the strong catalytic oxidation ability of the sample [47]. All three Co3O4-ZSM-5 samples displayed a small peak below 450 ℃, and MH-Co3O4@ZSM-5 showed a slightly stronger and wider peak, which indicated that MH-Co3O4@ZSM-5 had a higher number and mobility of oxygen species, contributing to better catalytic activity. This result was in accordance with the XPS observations stated above.
The surface acid properties of samples were measured using NH3-TPD. It is reported that the surface acidic sites of the catalyst could provide centers for Cl-containing molecules adsorption [48]. As shown in Fig. 3c, three distinct desorption peaks were observed for the pristine ZSM-5 catalyst spectrum, while no obvious peak was observed in the spectrum of pristine Co3O4. One ammonia desorption peak was observed for the three Co3O4-ZSM-5 samples, which indicated that the NH3 desorption peaks in the spectra of the three Co3O4-ZSM-5 samples were mainly caused by the acidity of ZSM-5. Obviously, the NH3 desorption peak in the spectrum of the MH-Co3O4@ZSM-5 catalyst was larger than that in the spectra of the other two catalysts in the CH sample ranking last. It was because the microwave and dynamic routes ensured the rapid uniform heating and contact of the reactants which increased the ZSM-5 yield.
According to the NH3-DRIFT profiles (Fig. 3d), the peaks in the range of 1850−1640 cm−1 and 1500−1400 cm−1 could be ascribed to NH4+ species on Brønsted acid sites [49, 50]. The bands at 1258, 1238, 1183, and 1136 cm−1 were assigned to NH3 adsorbed on Lewis acid sites [51, 52]. ZSM-5 and Co3O4-ZSM-5 samples all obtained peaks attributed to Lewis acid sites and Brønsted acid sites [53], indicating that they all obtained both Brønsted and Lewis acid sites.
The catalytic oxidation of DCM was carried out to evaluate the catalytic performance of the catalysts. Fig. 4 depicts the conversion of DCM and mineralization rate over pristine Co3O4, ZSM-5, and the three Co3O4-ZSM-5 samples synthesized by different methods. The pristine Co3O4 showed the best activity before 335 ℃. The mineralization rate of the Co3O4 sample was lower than that of Co3O4-ZSM-5 catalysts and only reached 62.5% at 435 ℃. ZSM-5 showed the lowest mineralization rate, and its activity was the lowest after 285 ℃. Among all the Co3O4-ZSM-5 samples, the MH-Co3O4@ZSM-5 showed the best activity, and DCM conversion reached 99.7% at 397 ℃. Moreover, it also obtained the highest mineralization rate over 99.0% at 397 ℃. The activity of CH-Co3O4/ZSM-5 was the lowest before 295 ℃ and increased quickly as the temperature went up with T50 of 332 ℃ and T90 of 404 ℃. The mineralization rate of the CH sample was a little higher than that of the DH sample. The sample synthesized by the dynamic hydrothermal method showed an activity with a T50 of 350 ℃ and T90 of 422 ℃. An irregular increase of DCM conversion occurred with a plateau for DH-Co3O4@ZSM-5 catalyst. It could be assigned to the deactivation/poisoning of the sample during the reaction [7, 54]. The mineralization rate of the DH catalyst was the lowest among all the Co3O4-ZSM-5 samples. The activity of the samples was in the order of MH-Co3O4@ZSM-5 > CH-Co3O4/ZSM-5 > DH-Co3O4@ZSM-5 according to T90 values. The substantial differences in activity test revealed that the preparation method was a key factor in obtaining highly active Co3O4-ZSM-5 catalysts. Combining the characterization results above, the main reasons why the MH sample showed the best activity included more surface adsorbed oxygen species, higher ratio of Co3+/Co2+, greater reducibility, oxygen mobility, and acidity. The reason why CH sample was more active at high temperature than the DH sample was that it had a larger specific surface area and higher ratio of Co3+/Co2+.

|
Download:
|
| Fig. 4. Light-off curves of (a) DCM catalytic degradation, (b) mineralization rate, and (c) byproduct yields over the tested samples. Reaction conditions: [CH2Cl2] =1000 ppm, [O2] = 10%, balanced N2, catalyst 0.2 g, and GHSV =30, 000 h−1. | |
{kind=link}
Although Co3O4 catalysts exhibited best DCM conversion 335 ℃, more byproducts were produced during the reaction. As shown in Fig. 4c, Co3O4 obtained the highest byproduct yield in the reaction process and reached 44.0% at 351 ℃. The by-products yield over ZSM-5 was higher than three Co3O4-ZSM-5 samples as well, ranking first at 434 ℃. For Co3O4-ZSM-5 samples, byproduct yields were less than 15%. Therefore, the combination of ZSM-5 and Co3O4 led to fewer byproducts. The byproduct distribution would be discussed later in this paper.
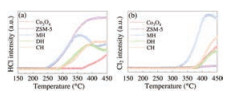
The dechlorination ability is of great important to the catalytic oxidation of CVOCs [55]. TPSR experiments equipped with mass spectrometry (MS) were carried out (Fig. 5) to investigate the changes in HCl and Cl2 during oxidation process over the samples. It was found that Brønsted acid sites were conducive to the rapid removal of HCl because the surface hydroxyl group of Brønsted acid sites could serve as the H resource for HCl formation [7]. For ZSM-5, HCl desorption appeared at around 247 ℃ which was attributed to the abundant Brønsted acid sites. Negligible Cl2 desorption was recorded for ZSM-5, indicating the absence of the Deacon reaction (HCl+O2 → Cl2 + H2O) [42]. HCl desorption on Co3O4 appeared at around 364 ℃ and Cl2 at 384 ℃ because Co3O4 was active for the Deacon reaction [19]. The appearance of HCl was observed at 240, 274, and 288 ℃ for the MH, CH, and DH samples, respectively, and Cl2 was at 320, 363 and 376 ℃. Less HCl was detected in the Co3O4-ZSM-5 samples than ZSM-5 because Co3O4 or the synergistic effect of ZSM-5 and Co3O4 facilitated the Deacon reaction and led to the rapid removal of Cl2 [56]. For the MH-Co3O4@ZSM-5 catalyst, HCl or Cl2 was detected first, and the desorption amount of HCl or Cl2 was the most among the three Co3O4-ZSM-5 catalysts. The results indicated that the MH sample was efficient for the formation and desorption of HCl and Cl2.

|
Download:
|
| Fig. 5. MS signals of (a) HCl and (b) Cl2 during DCM-TPSR experiments over tested samples. Reaction conditions: [CH2Cl2] =1000 ppm, [O2] = 10%, balanced He, catalyst 0.1 g, and GHSV = 30, 000 h−1. | |
{kind=link}
The generation of CH3Cl, CHCl3, and CCl4 during the TPSR experiments were shown in Fig. S2 (Supporting information). The distributions of CHCl3 and CCl4 were illustrated in Figs. S2a and S2b. Few polychlorinated products were produced over ZSM-5 and Co3O4-ZSM-5 samples during the process which was due to the Lewis acid sites. As for Co3O4, CCl3 and CCl4 were detected and reached their maximum at about 370 ℃ and 365 ℃, corresponding to the byproduct yields. It is reported that Co3O4 obtain high chlorination activity [16]. Therefore, CHCl3 and CCl4 might be produced by radical substitution over Co3O4 catalysts (CH2Cl2 + Cl2 → CHCl3 + HCl; CHCl3 + Cl2 → CCl4 + HCl) [57]. For Co3O4-ZSM-5 catalysts, few CH3Cl was detected (Fig. S3c in Supporting information) while ZSM-5 catalysts produced the most CH3Cl due to their poor reducibility [58].
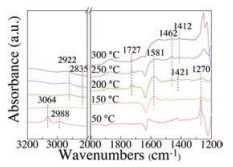
To further study the catalytic behaviors of the catalytic reaction, in situ DRIFT test was carried out on MH-Co3O4@ZSM-5 catalyst between 50−300 ℃ with the stream of 1000ppm DCM and balance He. As shown in the Fig. 6, there appeared several bands at 2919–3064 cm−1 were ascribed to C-H stretching vibrations, confirming the existence of different types of C-H bonds [59]. The bands at 3064, 2988 and 1270 cm−1 were assigned to the adsorbed DCM on the surface of the catalyst [60]. The bands at 2835–2922 cm−1 were assigned to methyl antisymmetric stretching and symmetric stretching of absorbed methoxy species [61]. While the other bands at 1412–1421 cm−1 refer to the C-H vibrations of surface dioxymethylene species [59, 62]. The bands generated at 1727 cm−1 over 200 ℃ refered to the C=O vibrations of formaldehyde [62]. The bands at around 1581 cm−1 was assigned to the asymmetric stretching of formate species [59]. The results of in situ DRIFT confirmed that methoxy species, formaldehyde, and formate were the main intermediates of the reaction.

|
Download:
|
| Fig. 6. In situ DRIFT spectra over MH-Co3O4@ZSM-5catalyst with the stream of 1000 ppm DCM/He. | |
{kind=link}
Here, a pathway over MH-Co3O4@ZSM-5 catalyst was deduced: (1) DCM molecule adsorbed on acidic sites or oxygen vacancies of the catalyst. (2) After the rupture of C–Cl bonds, HCl, chloromethoxy species and dioxymethylene species were generated. HCl could be further converted to Cl2 via the Deacon reaction. (3) The chloromethoxy species could be subjected to attack by OH groups and lattice oxygen to form formaldehyde [63] and further oxidized into formate species. Dioxymethylene species could also be oxidized into formate species, or form the mixture of -COOH and CH3O- species simultaneously by Cannizzaro type disproportionation [64]. (4) The intermediates above were further oxidation by surface active oxygen species to form CO, CO2 and H2O.
In summary, three different Co3O4-ZSM-5 catalysts were prepared by the microwave hydrothermal method, dynamic hydrothermal method, and conventional hydrothermal method for the catalytic decomposition of dichloromethane. Only the microwave hydrothermal process successfully obtained uniform core-shell structure catalysts (MH-Co3O4@ZSM-5). The MH sample showed the best catalytic activity, mineralization, and dechlorination ability for DCM. Further characterizations over the samples showed that MH-Co3O4@ZSM-5 exhibited the most surface adsorbed oxygen species, the highest ratio of Co3+/Co2+, abundant acidic sites, greater reducibility, oxygen mobility, and best HCl and Cl2 desorption ability among the tested Co3O4-ZSM-5 catalysts, which were beneficial for DCM oxidation. The combination of Co3O4 and ZSM-5 could effectively reduce the generation of by-products (such as CH3Cl, CHCl3, and CCl4). Methoxy species, formaldehyde, and formate were the main intermediates of the reaction and a reaction mechanism for the catalytic oxidation of DCM over the MH sample was proposed. Different hydrothermal synthetic methods influence the physicochemical characteristics of catalysts. Hence, the present work exhibits good potential for the rational design and synthesis of core-shell catalysts for CVOCs oxidation.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 51708492), National Key Research and Development Plan of China (No. 2016YFC0204700), Zhejiang Provincial "151" Talents Program, and the Program for Zhejiang Leading Team of S & T Innovation (No. 2013TD07).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.09.031.
| [1] |
M.C. Krol, J. Lelieveld, D.E. Oram, et al., Nature 421 (2003) 131-135. DOI:10.1038/nature01311 |
| [2] |
M.J. Moran, J.S. Zogorski, P.J. Squillace, Environ. Sci. Technol. 41 (2007) 74-81. DOI:10.1021/es061553y |
| [3] |
K. Sheng, H.F. Lu, A.B. Sun, et al., Chin. Chem. Lett. 30 (2019) 895-898. DOI:10.1016/j.cclet.2019.01.027 |
| [4] |
S. Pitkäaho, S. Ojala, T. Kinnunen, R. Silvonen, R.L. Keiski, Top. Catal. 54 (2011) 1257-1265. DOI:10.1007/s11244-011-9748-0 |
| [5] |
X. Zhang, Y.X. Liu, J.G. Deng, et al., Appl. Catal. B: Environ. 257 (2019) 12. |
| [6] |
J.J. Liu, X.X. Dai, Z.B. Wu, X.L. Weng, Chin. Chem. Lett. 31 (2020) 1410-1414. DOI:10.1016/j.cclet.2020.03.056 |
| [7] |
Q.G. Dai, Z.Y. Zhang, J.R. Yan, et al., Environ. Sci. Technol. 52 (2018) 13430-13437. DOI:10.1021/acs.est.8b05002 |
| [8] |
E. Lynge, A. Anttila, K. Hemminki, Cancer Causes Control 8 (1997) 406-419. DOI:10.1023/A:1018461406120 |
| [9] |
A.A. Meharg, D. Osborn, Nature 375 (1995) 353-354. DOI:10.1038/375353a0 |
| [10] |
S.Y. Kang, M. Wang, N. Zhu, et al., Chin. Chem. Lett. 30 (2019) 1450-1454. DOI:10.1016/j.cclet.2019.03.023 |
| [11] |
J.I. Gutierrez-Ortiz, B. de Rivas, R. Lopez-Fonseca, S. Martin, J.R. GonzalezVelasco, Chemosphere 68 (2007) 1004-1012. DOI:10.1016/j.chemosphere.2007.02.025 |
| [12] |
Y.Q. Wang, Y.F. Xue, C.C. Zhao, et al., Chem. Eng. J. 300 (2016) 300-305. DOI:10.1016/j.cej.2016.04.007 |
| [13] |
P. Yang, S. Yang, Z. Shi, Z. Meng, R. Zhou, Appl. Catal. B: Environ. 162 (2015) 227-235. DOI:10.1016/j.apcatb.2014.06.048 |
| [14] |
P. Yang, J. Li, L.F. Bao, et al., Chem. Eng. J. 361 (2019) 1400-1410. DOI:10.1016/j.cej.2018.10.071 |
| [15] |
B. de Rivas, R. Lopez-Fonseca, C. Jimenez-Gonzalez, J.I. Gutierrez-Ortiz, Chem. Eng. J. 184 (2012) 184-192. DOI:10.1016/j.cej.2012.01.027 |
| [16] |
T. Cai, H. Huang, W. Deng, et al., Appl. Catal. B: Environ. 166 (2015) 393-405. DOI:10.1016/j.apcatb.2014.10.047 |
| [17] |
B. de Rivas, R. Lopez-Fonseca, C. Jimenez-Gonzalez, J.I. Gutierrez-Ortiz, J. Catal. 281 (2011) 88-97. DOI:10.1016/j.jcat.2011.04.005 |
| [18] |
J. Gonzalez-Prior, R. Lopez-Fonseca, J.I. Gutierrez-Ortiz, B. de Rivas, Appl. Catal. B: Environ. 199 (2016) 384-393. DOI:10.1016/j.apcatb.2016.06.046 |
| [19] |
Y.J. Lao, N.X. Zhu, X.X. Jiang, et al., Catal. Sci. Technol. 8 (2018) 4797-4811. DOI:10.1039/C8CY01484A |
| [20] |
Q.G. Dai, Q. Zhu, Y. Lou, X.Y. Wang, J. Catal. 357 (2018) 29-40. DOI:10.1016/j.jcat.2017.09.022 |
| [21] |
Y. Cheng, J.S. Li, L.J. Wang, X.Y. Sun, X.D. Liu, Sep. Purif. Technol. 51 (2006) 210-218. DOI:10.1016/j.seppur.2006.02.002 |
| [22] |
A. Aranzabal, M. Romero-Saez, U. Elizundia, J.R. Gonzalez-Velasco, J.A. Gonzalez-Marcos, J. Catal. 296 (2012) 165-174. DOI:10.1016/j.jcat.2012.09.012 |
| [23] |
D. Divakar, M. Romero-Saez, B. Pereda-Ayo, et al., Catal. Today 176 (2011) 357-360. DOI:10.1016/j.cattod.2010.11.065 |
| [24] |
N. Blanch-Raga, A.E. Palomares, J. Martinez-Triguero, S. Valencia, Appl. Catal. B: Environ. 187 (2016) 90-97. DOI:10.1016/j.apcatb.2016.01.029 |
| [25] |
S. Askari, A.B. Siahmard, R. Halladj, S.M. Alipour, Powder Technol. 301 (2016) 268-287. DOI:10.1016/j.powtec.2016.06.018 |
| [26] |
J. Liu, D.F. Xue, Adv. Mater. 20 (2008) 2622-2627. DOI:10.1002/adma.200800208 |
| [27] |
K. Yao, S. Liu, Y.Y. Dong, et al., Mater. Design 90 (2016) 129-136. DOI:10.1016/j.matdes.2015.10.121 |
| [28] |
G. Yang, S.J. Park, Materials 12 (2019) 1-18. |
| [29] |
C.O. Kappe, Angew. Chem. Int. Ed. 43 (2004) 6250-6284. DOI:10.1002/anie.200400655 |
| [30] |
T.H.T. Vu, H.T. Au, L.T. Tran, et al., J. Mater. Sci. 49 (2014) 5617-5625. DOI:10.1007/s10853-014-8274-4 |
| [31] |
J.H. Yao, L.T. Lv, C.Q. Shen, et al., Ceram. Int. 39 (2013) 3359-3364. DOI:10.1016/j.ceramint.2012.07.093 |
| [32] |
C. Wang, W.C. Hua, G.T. Chai, C.H. Zhang, Y.L. Guo, Catalysts 9 (2019) 1-10. |
| [33] |
M. Ammar, Y. Cao, P. He, et al., Chin. Chem. Lett. 28 (2017) 1583-1589. DOI:10.1016/j.cclet.2017.03.015 |
| [34] |
Q.G. Dai, S.X. Bai, Y. Lou, et al., Nanoscale 8 (2016) 9621-9628. DOI:10.1039/C6NR01800A |
| [35] |
S.N. Xiao, W. Zhu, P.J. Liu, et al., Nanoscale 8 (2016) 2899-2907. DOI:10.1039/C5NR07589K |
| [36] |
C. Wang, C.H. Zhang, W.C. Hua, et al., RSC Adv. 6 (2016) 99577-99585. DOI:10.1039/C6RA18503G |
| [37] |
S.D. Li, S.P. Mo, J.Q. Li, H.D. Liu, Y.F. Chen, RSC Adv. 6 (2016) 56874-56884. DOI:10.1039/C6RA08394C |
| [38] |
B.Y. Bai, H. Arandiyan, J.H. Li, Appl. Catal. B: Environ. 142 (2013) 677-683. DOI:10.1016/j.apcatb.2013.05.056 |
| [39] |
G.Y. Long, M.X. Chen, Y.J. Li, et al., Chem. Eng. J. 360 (2019) 964-973. DOI:10.1016/j.cej.2018.07.091 |
| [40] |
Y. Yang, H. Li, H.T. Zhao, et al., J. Hazard. Mater. 371 (2019) 156-164. DOI:10.1016/j.jhazmat.2019.02.103 |
| [41] |
Z. Han, Y.X. Liu, J.G. Deng, et al., Catal. Today 327 (2019) 246-253. DOI:10.1016/j.cattod.2018.04.042 |
| [42] |
Y.M. Jiao, X. Chen, F. He, S.T. Liu, Chem. Eng. J. 372 (2019) 107-117. DOI:10.1016/j.cej.2019.04.118 |
| [43] |
Q.M. Ren, Z.T. Feng, S.P. Mo, et al., Catal. Today 332 (2019) 160-167. DOI:10.1016/j.cattod.2018.06.053 |
| [44] |
S.H. Xie, J.G. Deng, S.M. Zang, et al., J. Catal. 322 (2015) 38-48. DOI:10.1016/j.jcat.2014.09.024 |
| [45] |
J.K. He, S.Y. Chen, W.X. Tang, et al., Appl. Catal. B: Environ. 255 (2019) 11. |
| [46] |
Q.M. Ren, S.P. Mo, R.S. Peng, et al., J. Mater. Chem. A 6 (2018) 498-509. DOI:10.1039/C7TA09149D |
| [47] |
C.Y. Ma, Z. Mu, J.J. Li, et al., J. Am. Chem. Soc. 132 (2010) 2608-2613. DOI:10.1021/ja906274t |
| [48] |
S.X. Chen, Y. Wang, A.P. Jia, et al., Appl. Surf. Sci. 307 (2014) 178-188. DOI:10.1016/j.apsusc.2014.04.012 |
| [49] |
J. Su, W.Y. Yao, Y. Liu, Z.B. Wu, Appl. Surf. Sci. 396 (2017) 1026-1033. DOI:10.1016/j.apsusc.2016.11.083 |
| [50] |
L.A. Chen, J.H. Li, M.F. Ge, Environ. Sci. Technol. 44 (2010) 9590-9596. DOI:10.1021/es102692b |
| [51] |
H.K. Matralis, M. Ciardelli, M. Ruwet, P. Grange, J. Catal. 157 (1995) 368-379. DOI:10.1006/jcat.1995.1302 |
| [52] |
B. Thirupathi, P.G. Smirniotis, Appl. Catal. B: Environ. 110 (2011) 195-206. DOI:10.1016/j.apcatb.2011.09.001 |
| [53] |
W.L. Wang, Q.J. Meng, Y.H. Xue, et al., J. Catal. 366 (2018) 213-222. DOI:10.1016/j.jcat.2018.07.022 |
| [54] |
Q.G. Dai, S.X. Bai, J.W. Wang, et al., Appl. Catal. B: Environ. 142 (2013) 222-233. DOI:10.1016/j.apcatb.2013.05.026 |
| [55] |
J. Li, P. Zhao, S. Liu, Appl. Catal. A: Gen. 482 (2014) 363-369. DOI:10.1016/j.apcata.2014.06.013 |
| [56] |
Q.G. Dai, W. Wang, X.Y. Wang, G.Z. Lu, Appl. Catal. B: Environ. 203 (2017) 31-42. DOI:10.1016/j.apcatb.2016.10.009 |
| [57] |
G.A. Olah, B. Gupta, M. Farina, et al., J. Am. Chem. Soc. 107 (1985) 7097-7105. DOI:10.1021/ja00310a057 |
| [58] |
Q.G. Dai, J.Y. Wu, Q. Wu, et al., Appl. Catal. B: Environ. 249 (2019) 9-18. DOI:10.1016/j.apcatb.2019.02.065 |
| [59] |
Z. Zhang, H.Q. Xia, Q.G. Dai, X.Y. Wang, Appl. Catal. A: Gen. 557 (2018) 108-118. DOI:10.1016/j.apcata.2017.12.003 |
| [60] |
Y. Yang, S. Liu, H. Zhao, et al., Chemosphere 214 (2018) 553-562. |
| [61] |
J. Mei, S.J. Zhao, W.J. Huang, Z. Qu, N.Q. Yan, J. Hazard. Mater. 318 (2016) 1-8. DOI:10.1016/j.jhazmat.2016.06.010 |
| [62] |
G.Y. Popova, T.V. Andrushkevich, Y.A. Chesalov, E.S. Stoyanov, React. Kinet. Catal. Lett. 41 (2000) 805-811. |
| [63] |
Q.G. Dai, S.X. Bai, H. Li, et al., Appl. Catal. B: Environ. 168 (2015) 141-155. |
| [64] |
S. Cao, X.Q. Fei, Y.X. Wen, et al., Appl. Catal. A: Gen. 550 (2018) 20-27. DOI:10.1016/j.apcata.2017.10.006 |