 2021, Vol. 32
2021, Vol. 32 


b Zhejiang Provincial Engineering Research Center of Industrial Boiler & Furnace Flue Gas Pollution Control, Hangzhou 310058, China
Environmental catalysis is of great importance in air pollution control, which converts the air pollutants into harmless products via a range of heterogeneous catalytic reactions [1]. Typical examples include selective catalytic reduction (SCR) of NOx [2-4], catalytic destruction of organic wastes [5], the methane catalytic reforming with carbon dioxide [6], etc. Recent development in the environmental catalysis has been greatly accelerated by the increasingly stringent emission standards, while numerous techniques have been oriented to industrial-scale applications, making an important contribution to the improvement of air quality in China. As the core of environmental catalysis, rational design of environmental catalysts with an aim to maximize their catalytic activities have been extensively explored, which yields significant outcomes in terms of increasing the number of active sites [7, 8] and enhancing the redox ability of catalysts [9-11]. However, since most of environmental heterogeneous reactions involve two or more reactants, the pollutant destruction efficiency is not only dependent on the intrinsic properties of applied catalysts, but also on the mass transfer and collision probability of these multi-reactants. Current works put great efforts on modifying the catalyst intrinsic properties, while investigations onto how to regulate the reactants mass transfer rate has been paid much less attention; the latter is believed to play crucial role in determining the pollutant conversion efficiency and reaction selectivity.
Chlorinated volatile organic compounds (Cl-VOCs) are well-known with inherent bioaccumulation and potential carcinogenicity, many of which have been listed as priority control pollutants worldwide [12, 13]. Catalytic destruction of chlorinated organics remains a great challenge in environmental catalysis, owning to it encounters problems of catalyst deactivation [14] and secondary pollution (i.e., abundant more toxic byproducts) [15, 16], which severely hinders this technique towards industrial scale application [17-19]. This process is initiated by the scission of C–Cl bond at acidic (Brönsted/Lewis) sites or superficial oxygen vacancy and the activation of gaseous O2 at oxygen vacancy, followed by the reaction between multi-adsorbates to convert the Cl-VOCs into CO2, H2O, HCl/Cl2 and intermediates [20, 21]. The involvement of Cl-VOCs and O2 adsorptions at various active sites and the abundant reaction byproducts make the catalytic destruction of Cl-VOCs much ideal for exploring the importance of mass transfer in determining the pollutant destruction efficiency and product selectivity.
Herein, we choose CeO2 nanorods as a model catalyst, because it has abundant superficial oxygen vacancies [22, 23] that could provide sufficient adsorption sites for both of the Cl-VOCs and gaseous O2. Chlorobenzene (CB) was selected as typical Cl-VOCs, the oxidation of which has shown to easily generate reaction intermediates [24], and can be used to evaluate the reaction selectivity.
Furthermore, to get a contrasted catalyst, a HZSM-5 zeolite with abundant Brönsted acidic sites was introduced by using a dry-mixing route in a ball miller. This catalyst was expected to provide separated adsorption sites for Cl-VOCs and gaseous O2, as the Cl-VOCs were shown to preferentially adsorb on the Brönsted HZSM-5 sites. The separated adsorptions of Cl-VOCs and O2 and the poor-mixing of CeO2 and HZSM-5 effectively increased the mass transfer distance of their adsorbates, which should yield varied catalytic performance in comparison with their co-adsorption on the CeO2 vacancies.
The reaction characteristics and byproducts generation of CeO2 and CeO2/HZSM-5 catalysts in the catalytic CB oxidation (CBCO) were evaluated using a range of analytical techniques, including powder X-ray diffraction (XRD), transmission electron microscopy (TEM), temperature program reduction of hydrogen (H2-TPR), temperature program desorption of oxygen (O2-TPD), fourier transform infrared spectroscopy (FT-IR), gas chromatography mass spectrometry (GC-MAS), etc. XRD indicated the dry-mixing did not change the crystal structure of CeO2 and HZSM-5 (Fig. S1 in Supporting information). The former exhibited characteristic patterns at 28.7°, 33.1°, 47.4°, 56.3°, 69.7° and 76.9° with a cubic fluorite structure (JCPDS No. 89-8436), and the latter revealed an MFI type framework at 7.9°, 8.8°, 23.0°, 23.9°, 29.8°, 45.5° and 55.1° (JCPDS No. 44-0002). Scanning electron microscope (SEM) revealed that CeO2 was composed of monodispersed nanorods (200−500nm in length) and in the CeO2/HZSM-5, these nanorods were much shorter (50−200nm) and showed certain agglomerations (Fig. S2 in Supporting information). Energy dispersive X-ray spectroscopy (EDX) mapping indicated the HZSM-5 and CeO2 were not well mixed, owning to the use of dry mixing method (Fig. S3 in Supporting information). The Brunauer-Emmet-Teller (BET) surface area measurements showed the CeO2 with a surface area of 96.0m2/g, which was lower than that of CeO2/HZSM-5 (120.1m2/g), attributing to the HZSM-5 with a high BET surface area of 180.1m2/g.
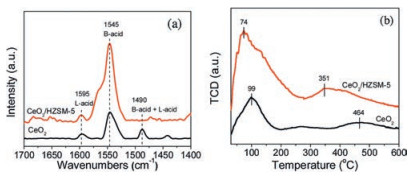
To confirm the existence of Brönsted acidity in the CeO2/HZSM-5 catalyst, pyridine adsorption infrared spectroscopy (Py-IR) and NH3 temperature programmed desorption (TPD) were conducted. As shown in Fig. 1a, the pyridine desorption peaks mainly located at 1595, 1545, and 1490 cm−1, which correspond to the Lewis acidic site, Brönsted acidic site and the combination of them, respectively [25, 26]. In comparison with CeO2, the CeO2/HZSM-5 catalyst exhibited a very intense peak at 1545 cm−1, suggesting that the introduction of HZSM-5 greatly enhanced the Brönsted acidity of the catalyst. This acidity was mainly derived from the proton H on the surface of HZSM-5. The amounts of acidic sites were also greatly increased by introducing the HZSM-5. In the NH3-TPD profile, the type of acids can be divided into weak acid (below 200 ℃), medium strong acid (200−400 ℃), and strong acid (above 400 ℃) based on the NH3 desorption temperature. As shown in Fig. 1b, the CeO2 exhibited two broad NH3 desorption peaks centered at 99 ℃ and 464 ℃, both of which were resulted from the Ce4+/Ce3+ (dominant) and the surface acidic hydroxyl group (bridged OHad) [27]. After loading the HZSM-5, the intensity of NH3 desorption peaks were significantly enhanced, and shifted to 74 ℃ and 351 ℃, respectively, suggesting that enriched weak and medium strong acidities were introduced to the CeO2/HZSM-5 catalyst, consistent with the Py-IR results.

|
Download:
|
| Fig. 1. (a) pyridine-IR and (b) NH3-TPD profiles of CeO2 and CeO2/HZSM-5 catalysts. | |
{kind=link}
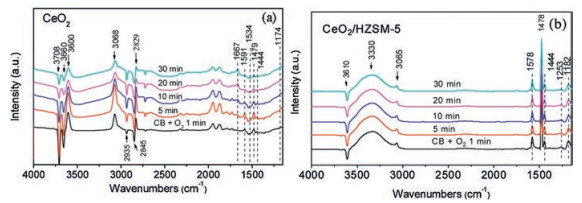
The selective adsorption of CB on the CeO2 and CeO2/HZSM-5 catalysts were confirmed using in situ FT-IR analyses. The spectra were collected at 150 ℃ in a stream of 500ppm CB and 10vol% O2 within 30min. As shown in Fig. 2a, the bands at 1591, 1479 and 1444 cm−1 are assigned to C=C degenerate stretching vibrations of the aromatic ring [28]. According to the literature [29], on the dehydroxylated defect-free CeO2 surface, CB adsorption was mainly through Ce4+⋯π-electron type interaction, while on the hydroxylated surface, this preceded via a dual-site interaction (OH⋯π-electron and OH⋯Cl). During the preparation of CeO2 nanorods, a large number of hydroxyl groups remained on the catalyst surface after alcohol washing. As a result, the CB was shown to initially adsorb on the Ce–OH site. This is confirmed by the changes of −OH vibration, which exhibited inverted peaks in the range of 3750−3625 cm−1 after CB adsorption. The appearance of 3600 cm−1 band is considered as the result of the migration of these inverted peaks, owning to the disturbance of adsorbed species [29]. The bands in the range of 2000−1700 cm−1 can be attributed to the out of plane distortion harmonics (combination and overtones) of the C–H bond [30], which are derived from the interaction ofπ electron cloud of benzene ring and electron center of oxide surface [31]. The characteristic bands at 3068 and 2829 cm−1 are derived from the vibration of C–H on benzene ring [32]. These bands increased gradually in the first 10min, and then decreased, suggesting that the OH groups on the CeO2 surface were gradually consumed by CB adsorption.

|
Download:
|
| Fig. 2. In situ FT-IR spectra of (a) CeO2 and (b) CeO2/HZSM-5 catalysts at 150 ℃ in a stream of 500 ppm CB and 10 vol% O2 within 30 min. | |
{kind=link}
After 10min, a new band appeared at 1667 cm−1, which gradually increased with the measuring time. This band has been assigned to the CB adsorption on Ce3+-Vo sites [33], which could result in the cleavage of C–Cl band, leaving the Cl at oxygen vacancies (Vo). The dissociated Cl at the Vo is inclined to attack the C+ of phenyl, leading to an electrophilic chlorination and the formation of (poly) chlorinated byproducts [34]. The continued growth of this peak indicated that after the complete consumption of surface hydroxyls in the CeO2, the CB was mainly adsorbed on surface Vo sites. Additionally, the vibration bands at 1534 and 1174 cm−1 are assigned to the intermediate products of maleic acid [28] and the inverted bands at 2935 and 2845 cm−1 can be attributed to methylene (−CH2−) and methyl (−CH3) [29].Fig. 2b illustrates the adsorption of CB on the CeO2/HZSM-5 catalyst. It was noted that loading of the HZSM-5 effectively changed the adsorption model of CB on the catalyst surface, where the CB was found to mainly adsorb on the hydroxyls of HZSM-5, revealing the characteristic bands at 1578, 1478, 1444 and 1253 cm-1 [35]. Thein situ FT-IR analyses confirmed our assumption that the CB was preferentially adsorbed on the HZSM-5, which effectively separated the adsorption site with O2, while this separated adsorption model made the two adsorbates have a comparatively larger mass transfer distance than co-adsorbed on the CeO2.
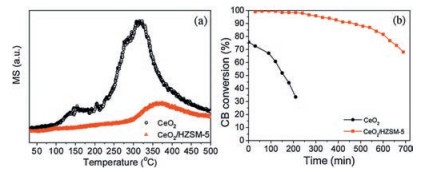
To investigate the reaction characteristics of CeO2 and CeO2/HZSM-5 in the CBCO reaction, a CB-TPSR experiment involving a flow of 500ppm CB and 10vol% O2 was conducted. The dynamic and timely generation of CO2 from this reaction were in situ monitored. As shown in Fig. 3a, the different mass transfer distance in the CeO2 and CeO2/HZSM-5 indeed resulted in a distinct change in CO2 generation, where the CeO2 with short transfer distance yielded an intense CO2 desorption peak in the temperature range of 225−450 ℃. In comparison, the CeO2/HZSM-5 with separated absorption sites of CB and O2 exhibited a much lower and postponed CO2 desorption peak. This result verifies that the mass transfer distance between the reactant adsorbates plays a crucial role in determining the CB destruction efficiency and CO2 selectivity, where short distance yielded much higher destruction efficiency and CO2 selectivity than the longer one. However, it was noted that the co-adsorption of CB and O2 at the Vo resulted in severe Cl poisoning of the catalyst, where in a 250 ℃ stability test (Fig. 3b), the CeO2 was shown to be rapidly deactivated, but the CeO2/HZSM-5 displayed a much better long-term stability. Since the introduction of HZSM-5 was shown to not significantly alter the redox properties of CeO2 catalyst (as confirmed by H2-TPR and O2-TPD analyses in Figs. S4-S5 in Supporting information), we believed that the higher long-term stability of CeO2/HZSM-5 should be attributed to the preferential adsorption of CB at HZSM-5 that hindered the Cl occupation at Vo and ensured the continuous O2 activation for CBCO reaction.

|
Download:
|
| Fig. 3. (a) CB-TPSR profiles of CO2 yield and (b) a stability test at 250 ℃ on the CeO2 and CeO2/HZSM-5 catalysts; Reaction conditions: GHSV = 10 000 mL g−1 h−1, 500 ppm CB, N2 flow rate =145 mL/min, O2 flow rate = 15 mL/min. | |
{kind=link}
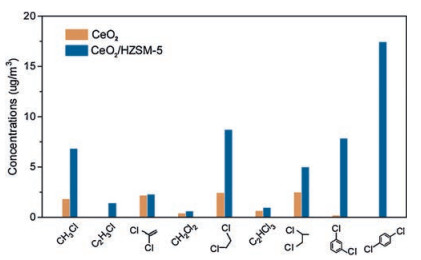
Reaction byproducts, particularly toxic polychlorinated organics in the off-gases were quantitatively analysed using a calibrated GC–MS system. As shown in Fig. 4, the CeO2 and CeO2/HZSM-5 catalysts both generated certain polychlorinated byproducts, including polychlorinated alkanes, polychlorinated alkenes and dichlorobenzenes, amongst which, the dichlorobenzenes should be paid the most concern as they are easily converted into dioxins, leading to severe secondary pollution to the environment [36-38]. The CeO2 yielded approximately 2μg/m3 of p-dichlorobenzene, while for the CeO2/HZSM-5, the amounts of m-dichlorobenzene were measured at 5μg/m3, and the p-dichlorobenzene was shown to be as high as 38μg/m3. Such a difference was show to originate from the excessive adsorption of CB on the HZSM-5 surface (Fig. S6 in Supporting information) that facilitated the electrophilic chlorination reaction. This reaction was assumed to precede through the electrophilic substitution of Cl over the Lewis acid sites of CeCl4 [39] that attacked the accumulated CB at Brönsted HZSM-5 sites, leading to the formation of dichlorobenzenes in the off-gas.

|
Download:
|
| Fig. 4. Quantitative analyses of polychlorinated byproducts collected in the 350 ℃ off-gases of CeO2 and CeO2/HZSM-5 catalysts. | |
{kind=link}
In summary, we have fabricated CeO2 and CeO2/HZSM-5 catalysts that were employed in the CBCO reaction to unveil the importance of reactant mass transfer in environmental catalysis. The co-adsorbed CB and O2 on the CeO2 surface resulted in a remarkably high CO2 generation, while those separately adsorbed on the CeO2/HZSM-5 yielded a much lower CO2 generation. This verifies our assumption that rational design of the mass transfer distance of reactant adsorbates can effectively regulate the pollutant conversion efficiency and product selectivity. The co-adsorption of CB and O2 was shown to cause severe deactivation of the CeO2 catalyst, as the dissociated Cl occupied the surface oxygen vacancy that hindered the O2 activation. While in the CeO2/HZSM-5, the CB was preferentially adsorbed on the Brönsted acidic sites of HZSM-5, which protected the oxygen vacancy from Cl poisoning, leading to a high long-term stability in CBCO reaction. However, the excessive adsorption of CB on the Brönsted sites distinctly promoted electrophilic chlorination reaction, which generated significant dichlorobenzenes in the off-gas, causing a severe secondary pollution to the environment. The work conducted herein unveils that the design of environmental catalysts needs to consider both of catalyst intrinsic property and reactant mass transfer, as they can both affect the pollutant conversion, product selectivity, reaction stability and secondary pollution. To date, modification of the reactant mass transfer has been paid much less attention in environmental catalysis. Such an investigation could pave a new way for the development of highly efficient catalysts for environmental pollution control.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was financially supported by the National Key R&D Program of China (No. 2016YFC0202200), the National Natural Science Foundation of China (Nos. 21777140, 21922607) and the Outstanding Youth Project of Zhejiang Natural Science Foundation (No. LR19E080004).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.09.001.
| [1] |
C. He, J. Cheng, X. Zhang, et al., Chem. Rev. 119 (2019) 4471-4568. DOI:10.1021/acs.chemrev.8b00408 |
| [2] |
M. Shelef, Chem. Rev. 95 (1995) 209-225. DOI:10.1021/cr00033a008 |
| [3] |
G. Busca, L. Lietti, G. Ramis, F. Berti, Appl. Catal B: Environ. 18 (1998) 1-36. DOI:10.1016/S0926-3373(98)00040-X |
| [4] |
L.P. Han, S.X. Cai, M. Gao, et al., Chem. Rev. 119 (2019) 10916-10976. DOI:10.1021/acs.chemrev.9b00202 |
| [5] |
J. Wang, A. Yoshida, P.F. Wang, et al., Appl. Catal B: Environ. 271 (2020) 118941. DOI:10.1016/j.apcatb.2020.118941 |
| [6] |
B. Abdullah, N.A.A. Ghani, D.V.N. Vo, J. Clean. Prod. 162 (2017) 170-185. DOI:10.1016/j.jclepro.2017.05.176 |
| [7] |
Y.S. Kang, Y. Lu, K. Chen, et al., Coord. Chem. Rev. 378 (2019) 262-280. DOI:10.1016/j.ccr.2018.02.009 |
| [8] |
H.X. Zhou, S.P. Wang, B.W. Wang, X.B. Ma, S.Y. Huang, Chin. Chem. Lett. 30 (2019) 775-778. DOI:10.1016/j.cclet.2018.10.005 |
| [9] |
X.W. Wang, W.Y. Jiang, R.Q. Yin, et al., J. Colloid Interface Sci. 574 (2020) 251-259. DOI:10.1016/j.jcis.2020.04.047 |
| [10] |
Y. Du, Y.B. Shen, Y.L. Zhan, et al., Chin. Chem. Lett. 28 (2017) 1746-1750. DOI:10.1016/j.cclet.2017.05.018 |
| [11] |
W. Yu, D. Wei, W. Yifu, G. Limin, I. Tatsumi, Mol. Catal. 459 (2018) 61-70. DOI:10.1016/j.mcat.2018.08.022 |
| [12] |
B.B. Huang, C. Lei, C.H. Wei, G.M. Zeng, Environ. Int. 71 (2014) 118-138. DOI:10.1016/j.envint.2014.06.013 |
| [13] |
P. Yang, S.S. Yang, Z.N. Shi, et al., Appl. Catal B: Environ. 162 (2015) 227-235. DOI:10.1016/j.apcatb.2014.06.048 |
| [14] |
J. Liu, X. Dai, Z. Wu, X. Weng, Chin. Chem. Lett. 31 (2020) 1410-1414. DOI:10.1016/j.cclet.2020.03.056 |
| [15] |
X.X. Dai, X.W. Wang, Y.P. Long, et al., Environ. Sci. Technol. 53 (2019) 12697-12705. DOI:10.1021/acs.est.9b05088 |
| [16] |
W.L. Wang, Q.J. Meng, Y.H. Xue, et al., J. Catal. 366 (2018) 213-222. DOI:10.1016/j.jcat.2018.07.022 |
| [17] |
X.L. Liu, L. Chen, T.Y. Zhu, R.L. Ning, J. Hazard. Mater. 363 (2019) 90-98. DOI:10.1016/j.jhazmat.2018.09.074 |
| [18] |
R.W. van den Brink, R. Louw, P. Mulder, Appl. Catal B: Environ. 16 (1998) 219-226. DOI:10.1016/S0926-3373(97)00076-3 |
| [19] |
W.Y. Jiang, Y.L. Yu, F. Bi, et al., Environ. Sci. Technol. 53 (2019) 12657-12667. DOI:10.1021/acs.est.9b04155 |
| [20] |
H.A. Miran, M. Altarawneh, Z.T. Jiang, et al., Catal. Sci. Technol. 7 (2017) 3902-3919. DOI:10.1039/C7CY01096F |
| [21] |
P.F. Sun, W.L. Wang, X.L. Weng, X.X. Dai, Z.B. Wu, Environ. Sci. Technol. 52 (2018) 6438-6447. DOI:10.1021/acs.est.7b06023 |
| [22] |
H. Huang, Q.G. Dai, X.Y. Wang, Appl. Catal B: Environ. 158 (2014) 96-105. |
| [23] |
S.Y. Zhao, S.P. Wang, Y.J. Zhao, X.B. Ma, Chin. Chem. Lett. 28 (2017) 65-69. DOI:10.1016/j.cclet.2016.06.003 |
| [24] |
X.L. Weng, Q.J. Meng, J.J. Liu, et al., Environ. Sci. Technol. 53 (2019) 884-893. DOI:10.1021/acs.est.8b04582 |
| [25] |
C.A. Emeis, J. Catal. 141 (1993) 347-354. DOI:10.1006/jcat.1993.1145 |
| [26] |
M.A. Makarova, K. Karim, J. Dwyer, Microporous Mesoporous Mater. 4 (1995) 243-246. DOI:10.1016/0927-6513(94)00093-B |
| [27] |
Q. Dai, Z. Zhang, J. Yan, et al., Environ. Sci. Technol. 52 (2018) 13430-13437. DOI:10.1021/acs.est.8b05002 |
| [28] |
J. Lichtenberger, M.D. Amiridis, J. Catal. 223 (2004) 296-308. DOI:10.1016/j.jcat.2004.01.032 |
| [29] |
M. Nagao, Y. Suda, Langmuir 5 (1989) 42-47. DOI:10.1021/la00085a009 |
| [30] |
M.A. Larrubia, G. Busca, Appl. Catal B: Environ. 39 (2002) 343-352. DOI:10.1016/S0926-3373(02)00116-9 |
| [31] |
G. Ramis, G. Busca, V. Lorenzelli, J. Electron Spectros. Relat. Phenom. 64-65 (1993) 297-305. |
| [32] |
M.A. Larrybia, A. Gutierrez-Alejandre, J. Ramirez, G. Busca, Appl. Catal. A: Gen. 224 (2002) 167-178. DOI:10.1016/S0926-860X(01)00769-4 |
| [33] |
H. Huang, Y.F. Gu, J. Zhao, X.Y. Wang, J. Catal. 326 (2015) 54-68. DOI:10.1016/j.jcat.2015.02.016 |
| [34] |
Y.F. Gu, T. Cai, X.H. Gao, et al., Appl. Catal B: Environ. 248 (2019) 264-276. DOI:10.1016/j.apcatb.2018.12.055 |
| [35] |
X.L. Weng, P.F. Sun, Y. Long, Q.J. Meng, Z.B. Wu, Environ. Sci. Technol. 51 (2017) 8057-8066. DOI:10.1021/acs.est.6b06585 |
| [36] |
S. Nganai, S.M. Lomnicki, B. Dellinger, Environ. Sci. Technol. 45 (2011) 1034-1040. DOI:10.1021/es102948f |
| [37] |
S. Nganai, B. Dellinger, S. Lomnicki, Environ. Sci. Technol. 48 (2014) 13864-13870. DOI:10.1021/es504253w |
| [38] |
M. Altarawneh, B.Z. Dlugogorski, E.M. Kennedy, J.C. Mackie, Prog. Energy Combust. Sci. 35 (2009) 245-274. DOI:10.1016/j.pecs.2008.12.001 |
| [39] |
P.F. Sun, W.L. Wang, X.X. Dai, X.L. Weng, Z.B. Wu, Appl. Catal. B: Environ. 198 (2016) 389-397. DOI:10.1016/j.apcatb.2016.05.076 |