 2021, Vol. 32
2021, Vol. 32 


b Fujian Engineering and Research Center of Clean and High-valued Technologies for Biomass, Xiamen 361102, China;
c School of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310058, China;
d Department of Paper and Bioprocess Engineering, State University of New York College of Environmental Science and Forestry, Syracuse, New York 13210, United States;
e State Key Laboratory of Applied Microbiology Southern China, Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Open Laboratory of Applied Microbiology, Guangdong Microbial Culture Collection Center (GDMCC), Guangdong Institute of Microbiology, Guangdong Academy of Sciences, Guangzhou 510070, China
Furfuryl alcohol (FFA), as a versatile chemical intermediate derived from lignocelluloses biomass, has been widely used for the chemical manufacture, such as casting/foundry resins, pharmaceuticals and lubricants [1]. Selective hydrogenation of furfural (FF) to furfuryl alcohol (FFA) is considered to be a very important industrial process [2]. In the latest decades, improving catalyst performance for selective hydrogenation of FF to FFA is an important subject and it has obtained wildly attention. The Cu-Cr catalyst have usually been used for producing FFA in industry, yet it usually bring about environmental concern owing to the high toxicity of Cr(Ⅵ) [3]. Besides many noble (Pd, Pt) supported catalysts [4], numerous effort have been taken to synthesize various Cr-free catalysts for FFA production with non-noble metals (Cu, Ni and Co) [5-7]. Especially for Cu-based catalysts, such as Cu-MgO [8] and Cu-MgO promoted with Co, Ca, or Al [6, 9], Cu-Zn mixed oxides [10], and other supported Cu-based catalysts [11] are most employed for the selective hydrogenation of FF to FFA. In addition, Ni-B, Co-B, and Ni-Co-B amorphous alloys also exhibited an excellent performance in selectivity hydrogenation of FF to FFA [12-14]. Nevertheless, these FFA production is significantly dependent on external H2, which consumed a large amount of petroleum or coal resources [15, 16].
Alternatively, formic acid, as a liquid hydrogen source, has attracted great interest due to its sustainability [17, 18]. However, only a few studies refer to the application of formic acid as hydrogen donor for the selective catalytic transfer hydrogenation (CTH) of FF to FFA owing to the instability of FF in acid environment. Au/meso-CeO2 obtained a 96% FFA yield from the CTH of FF with HCOOK as hydrogen donor [19]. Rh/ED-KIT-6 firstly achieved more than 97% FFA yield from the CTH of FF using formic acid as hydrogen donor, however this catalyst requires a very complex preparation process and also needs high-loading of noble metal (50.42 wt% Rh) [20]. In our previous study, in-situ prepared nano Cu-Pd/C catalyst exhibited the best performance with FFA yield of over 98.1%. Meanwhile, Cu species in Cu-Pd/C catalyst was also able to catalytically decompose formic acid to generate hydrogen and hydrogenate FF to FFA [21].
In the present study, the Cr-free supported catalysts, CuCs(x)-MCM, were prepared with varying ratio of Cu and Cs for the CTH of FF to FFA with formic acid as hydrogen donor. Cs was used as a promoter to Cu metal to enhance the activity of catalyst and selectivity of FFA. The effects of different reaction parameters, such as metal loading and different metal ratio, on the catalytic performance were investigated to optimize the FF conversion and FFA selectivity.
In order to clarify the function of Cu and Cs species, we design a series of experiments, which including contrast test of catalysts, stability test of catalyst, and a series of catalyst characterization. All the CTH of FF to FFA was performed in a 50 mL stainless steel Parr autoclave with a magnetic stirrer. In a typical run, 0.3 g FF, 20 mL 1, 4-dioxane, 0.575 g FA, and 0.04 g solid catalyst were charged into the reactor, and then the reactor was sealed. After purging with N2 for 3–5 times, the reactor was heated to the target temperature with stirring of 500 rpm for a desired time. After the reaction, the reactor was cooled to room temperature quickly. The gas and liquid samples were collected separately for subsequent analysis, and the catalyst was recovered for detection and recycling.
In a typical experiment, the catalysts were prepared by conventional incipient wetness impregnation method [22]. The detail information of materials, catalyst preparation, catalyst characterization method and products analyses were described in Supporting information. And some experiments results, which including catalyst screening, effect of catalyst loading on catalytic activity, correlation analysis of factors relative to catalytic activity, stability test of catalyst, were described in Supporting information. The following are the key results and discussion of the catalyst characterization.
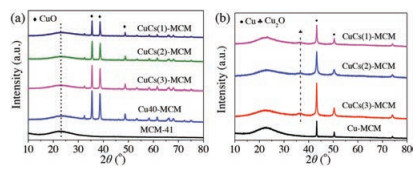
The X-ray diffraction (XRD) patterns of the calcined and in-situ reduced catalysts are shown in Fig. 1. As shown in Fig. 1a, MCM-41 usually exhibits characteristic peaks in the range of 2θ=2.2°–4.0°, thereby, almost no obvious characteristic peak of MCM-41 was detected between 10°–80° except a shoulder peak at 2θ=23.0°, which arose in all calcined catalysts. Other catalysts showed the characteristic peaks of CuO at 2θ=35.5°, 38.5°, 48.4°, respectively [23], indicating that CuO phase still remained unchanged with the introduction of Cs. Whereas, the intensity of the CuO characteristic peaks gradually decreased with the increasing Cs content. Moreover, the average size of the CuO particle became larger showed in Table S2 (Supporting information), demonstrating the incorporation of Cs species in CuO phase. In addition, the characteristic peak of Cs species was not detected for all catalysts, possibly being due to the formation of amorphous Cs species or the high dispersion of Cs species in CuO catalyst.

|
Download:
|
| Fig. 1. XRD patterns of the calcined (a) and in-situ reduced (b) catalysts. | |
{kind=link}
Fig. 1b shows the XRD pattern of the in-stiu reduced catalysts after the reaction at 170 ℃ for 1 h. The diffraction peaks presented in the XRD pattern were corresponded to Cu instead of CuO. Moreover, a barely observed diffraction peak of Cu2O detected at 2θ=36.2° testified that CuO was reduced completely, which is consistent with the previous work that Cu was produced at the beginning of the reaction from the complete reduction of CuO [21]. Furthermore, the introduction of Cs species made the intensity of the diffraction peaks of the reduced Cu particles higher and the peak broader, corresponding to smaller crystal size. According to the calculation by Scherrer formula (Table S2 in Supporting information), the size of Cu particle varies from 45.19 nm in Cu40-MCM to 20.92–26.08 nm in CuCS(x)-MCM, especially for CuCs(2)-MCM, which is probably brought about by the interaction between Cu and Cs [24].
To obtain a better understanding of reaction mechanisms, the properties of the support MCM-41 and several CuCS(x)-MCM catalysts were analyzed via N2 adsorption-desorption, and the results are presented in Table S2 (Supporting information). The SBET and pore diameter of the MCM-41 were 1060.58 m2/g and 3.72 nm, respectively, which favors the dispersion of active components. Some metal particles deposited in the pore structure of MCM-41 resulted in an obvious decrease in the SBET, pore volume (Vp) and pore diameter (Dp) of CuCs(x)-MCM. According to the N2O oxidation analysis, the dispersion (DCu) and specific surface area (SCu) of Cu were increased to some extent owing to the incorporation of Cs species into Cu-MCM (Table S2 in Supporting information).
The N2 adsorption-desorption isotherms of the support MCM-41 and CuCs(x)-MCM catalysts are shown in Fig. S1a (Supporting information). The N2 adsorption-desorption isotherms of all catalysts exhibited Langmuir type Ⅳ isotherms with a H1-type hysteresis loop, suggesting that these catalysts still retained ordered mesoporous structure similar to that of the support MCM-41 after the Cu and Cs species were introduced into MCM-41. This is the crucial property for the activity and stability of catalyst. Moreover, CuCs(x)-MCM exhibited a narrower pore size distribution for the introduction of Cs species (Fig. S1b in Supporting information), meaning that the pore size of the catalysts is quite uniform [25]. These results also confirm that the Cu and Cs species was successfully loaded on the surface of support MCM-41.
For further understand the morphology of catalyst, some samples was analyzed by Scanning Electronic Microscopy (SEM) and transmission electron microscopy (TEM). Fig. 2 shows the SEM image of the calcined and the in-situ reduced CuCs(2)-MCM. The smooth surface of the calcined CuCs(2)-MCM and no obvious metal particles were observed (Fig. 2a), which was similar to that of the MCM-41 support. However, uniform and fine metal particles were observed on the surface of in-situ reduced CuCs(2)-MCM (Fig. 2b). According to the previously study, the in-situ reduction of CuO in formic acid system involved two steps: firstly, CuO was dissolved into Cu2+, and then the Cu2+ was reduced to Cu [21]. Therefore, it is inferred that the visible metal particles on the catalyst surface are bimetallic nanoparticles. Furthermore, O, Si, Cu and Cs elements were detected from the randomly selected two particles on the in-situ catalyst by Energy dispersive Spectrometer (EDS) EDS element analysis (Fig. 2c). The relative content of these elements was relatively stable, signifying that the active element distribution is uniform in the catalyst prepared by in-situ reduction.

|
Download:
|
| Fig. 2. SEM image of the calcined (a) and in-situ reduced (b) CuCs(2)-MCM and EDS element analysis (c) for the in-situ reduced CuCs(2)-MCM. TEM images of the calcined (d) and in-situ reduced (e) CuCs(2)-MCM. | |
{kind=link}
In order to more clearly observe the morphology of the CuCs(2)-MCM catalyst, the calcined and the in-situ reduced CuCs(2)-MCM were analyzed by TEM test. As shown in Fig. 2d, the ordered network structure of support MCM-41 was clearly observed in the two different samples, suggesting a good thermal stability of the support during the preparation of the catalyst. Obvious particles ranged from 50 nm to 150 nm were also clearly observed on the surface of the support (Fig. 2e), which may be the active Cu particles modified by Cs species. This is consistent with the SEM analysis. In-situ CuCs(2)-MCM and the catalyst reused for 7 times were analyzed by TEM. The average diameter of Cu particles slightly increased from 35.1 nm in in-situ CuCs(2)-MCM to 45.8 nm in reused CuCs(2)-MCM reused for 7 times (Fig. S2 in Supporting information), which probably contributes to the slightly decrease in the yield of FFA after the catalyst was reused for 7 times. This result is consistent with the recyclability of CuCs(2)-MCM catalyst for the CTH of FF into FFA.
Then, we analyzed the catalysts with H2-Temperature Programmed Reduction (TPR). In the previous study, the reduction temperature of small CuO particles with good dispersion is lower than that of large particles [26]. As shown in Fig. S3 (Supporting information), Cu40-MCM without Cs species incorporation showed two reduction peaks at 250 ℃ and 193 ℃, respectively. While for CuCs(x)-MCM with different ratios of Cu: Cs, only one reduction peak with a narrow width was presented with decreased reduction temperature. It is reasonably inferred that the smaller CuO particles is formed due to the introduction of Cs species, improving the dispersion of CuO on the surface of MCM (Table S2 in Supporting information). Undoubtedly, interaction between Cu and Cs element did its work during calcination. This result is similar to that of the XRD analysis. It is also consistent with previous report that doping the reduction temperature of CuO is declined with the doping of metal elements [27, 28].
It is generally known that the surface acidity of Cu-based catalyst has great effect on the CTH of FF. Acidic site with strong oxophilic property on the surface of Cu-based catalyst contributes to the hydrogenolysis of saturated C-O bonds [29, 30], which is not beneficial for the hydrogenation of FF to FFA. Therefore, modifying the acidity of the Cu-based catalyst by doping other metal elements or loading on different supports facilitates the selective hydrogenation of FF to FFA. In this case, the acidity of the in-situ Cu-based catalysts were analyzed by NH3-Temperature Programmed Desorption (TPD). As shown in Fig. S4a (Supporting information), the pure Cu catalyst possessed both weak acid and strong acid sites, while the MCM-41 support had hardly any acidic sites. But Cu40-MCM presented a broad NH 3 desorption peak at high temperature centered at 504 ℃, showing a strong interaction between Cu and support MCM-41. Moreover, the acid distribution of the Cu-based catalyst is affected significantly by MCM-41 [29]. Once the Cs species was introduced into the Cu-MCM-41 catalyst, the NH3 desorption temperatures of CuCs(x)-MCM were significantly decreased (299-338 ℃) and the strong acidity was restrained. In general, the NH3 desorption temperature of CuCs(x)-MCM were variable, whereas CuCs(2)-MCM had the lowest one, indicating that the ratio of Cu: Cs also has great influence on the acid distribution on the surface of the catalyst (Fig. S4b in Supporting information).
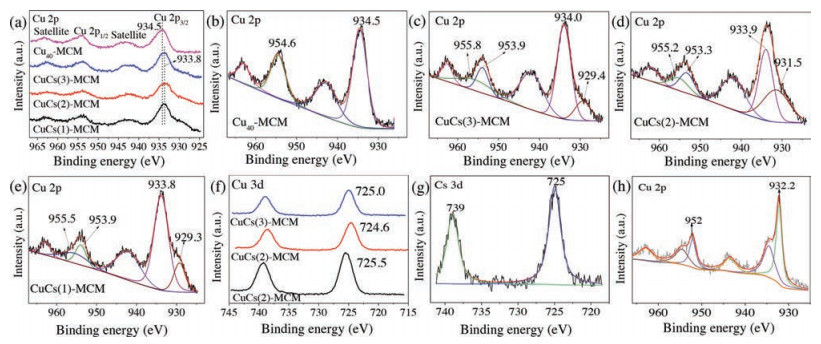
For further clarify the roles of Cu and Cs in the CTH of FF to FFA. The chemical states of Cu and Cs on the catalyst surface were analyzed by X-ray photoemission spectroscopy (XPS) characterization. The XPS spectra of the calcined catalysts CuCs(x)-MCM are presented in Fig. 3a. The photoelectron peaks around 934.5 eV (Cu 2p3/2) and 952.6 eV (Cu 2p1/2), as well as the characteristic satellite peaks in the range of 942.0–944.0 eV for all samples, showed that Cu element in the calcined CuCs(x)-MCM was in an oxide state. However, the deconvolution of Cu 2p3/2 in CuCs(x)-MCM presented two peaks, including the electron binding energy of Cu2+ and Cu0/+ between 933.8–934.0 eV and 929.3–931.5 eV, respectively. The electron binding energy of Cu2+ in CuCs(x)-MCM was significantly lower than that in Cu40-MCM (934.5 eV) owing to the doping of Cs (Figs. 3b–e), inferring a probable electron transfer from Cs to Cu [31, 32], which led to the decline in the surface acidity of the Cu catalyst. Especially, the reduced Cu0/+ observed in CuCs(x)-MCM suggested that Cs provided the electron transfer and effected the chemical state of Cu species. Compared with the standard electron binding energy of Cs (Binding energy of Cs 3d5/2 is 724.0 eV), the binding energy of Cs 3d5/2 shifted to 724.6–725.5 eV from 724.0 eV (Fig. 3f), further confirming an electronic effect between Cs and Cu. And the photoelectron peaks around 932.6 eV (Cu 2p3/2) and 952.6 eV (Cu 2p1/2), as well as the characteristic satellite peaks in the range of 942.0–944.0 eV for in-situ CuCs(x)-MCM, showed that Cu element in the in-situ CuCs(x)-MCM was in mental state (Fig. 3g). Compared with the standard electron binding energy of Cu (Binding energy of Cu 2p3/2 is 732.6 eV, Cu 2p1/2 is 752.6 eV), the binding energy of Cu 2p3/2 shifted to 732.2 eV from 732.6 eV, and the binding energy of Cu 2p1/2 shifted to 752 eV from 752.6 eV in in-situ CuCs(2)-MCM (Fig. 3g). And compared with the standard electron binding energy of Cs (Binding energy of Cs 3d5/2 is 724.0 eV), the binding energy of Cs 3d5/2 shifted to 725 eV from 724.0 eV (Fig. 3h), further confirming an electronic effect between Cs and Cu.

|
Download:
|
| Fig. 3. XPS spectra of Cu 2p (a), high-resolution photoelectron spectra of Cu 2p (b), (c), (d) and (e) of the calcined CuCs(x)-MCM catalysts and Cs 3d (f) of the calcined CuCs(x)-MCM catalysts, and high-resolution photoelectron spectrum of Cu 2p (g) and Cs 3d (h) of the in-situ CuCs(2)-MCM catalyst. | |
{kind=link}
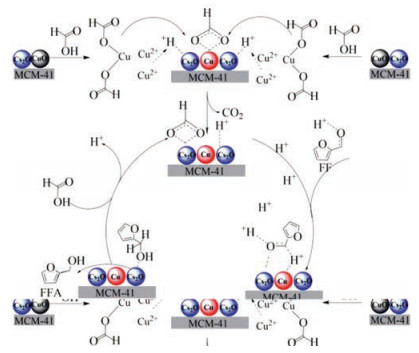
From the above analysis results, the possible reaction path of CTH of FF to FFA on the CuCs-MCM catalyst was summarized as following. Previous study suggests copper ion also can decompose formic acid to generate in-situ hydrogen [33] and further form copper hydride [34]. Therefore, based on the analyses in this study and previous studies on the hydrogenation of FF, the plausible reaction mechanism for the CTH of FF to FFA is proposed, as illustrated in Scheme 1. Initially, CuO is dissolved by FA to form Cu(HCOO)2, which is adsorbed on the surface of the Cs/MCM [21]. Then, the HCOO- species are decomposed by Cu2+ to form Cu2+-H species along with the release of CO2. Subsequently, the Cu2+ is in-situ reduced by Cu2+-H species to nano Cu, which uniformly distributes on the Cs-MCM surface and forms CuCs-MCM. In addition, lower valence state of copper favors the decomposition of formate [20], CuCs-MCM undertakes dual roles of decomposing formate and hydrogenating FF to FFA. FF is adsorbed by the Lewis acidic sites on the surface of the CuCs-MCM through carbonyl oxygen atom [33, 35]. Finally, the hydrogen atoms on the Cu-Cs alloys surface are transferred to the carbonylcarbon of FF, resulting in the formation of FFA.

|
Download:
|
| Scheme 1. The possible reaction path of CTH of FF to FFA on the CuCs-MCM catalyst. | |
{kind=link}
From all the results and discussion, we think we have demonstrated an efficient and extremely durable CuCs(2)-MCM catalyst for the selective CTH of furfural (FF) to furfuryl alcohol (FFA) using formic as hydrogen donor. CuCs(2)-MCM achieved a 99.6% FFA yield at an optimized reaction conditions of 170 ℃, 1 h. Cs species played a favorable role in enhancing the hydrogenation activity as a promoter by adjusting the surface acidity of Cu catalyst to an appropriate level. Moreover, the doping of Cs made the size of Cu particles smaller and improved the dispersion of the Cu species. The good hydrothermal stability and ordered mesoporous structure of MCM-41 support was essential to ensure the stability and reusability of CuCs-MCM. Correlation analysis showed that surface acidity was the primary factor to affect the catalytic activity of CuCs(x)-MCM compared with the dispersity, specific surface area and particle size of Cu species in CuCs-MCM.
Declaration of competing interestWe declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and company that could be construed as influencing the position presented in, or the review of, the manuscript entitled.
AcknowledgmentThis work was supported by the National Natural Science Fund of China (Nos. 21776234, 21978246).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.07.044.
| [1] |
Y. He, Y. Ding, C. Ma, et al., Green Chem. 19 (2017) 3844-3850. DOI:10.1039/C7GC01256J |
| [2] |
F.H. Isikgor, C.R. Becer, Polym. Chem. 6 (2015) 4497-4559. DOI:10.1039/C5PY00263J |
| [3] |
S. Srivastava, P. Mohanty, J.K. Parikh, et al., Chin. J. Catal. 36 (2015) 933-942. DOI:10.1016/S1872-2067(15)60870-1 |
| [4] |
Á. O'Driscoll, J.J. Leahy, T. Curtin, Catal. Today 279 (2017) 194-201. DOI:10.1016/j.cattod.2016.06.013 |
| [5] |
M. Audemar, C. Ciotonea, K. De Oliveira Vigier, et al., ChemSusChem 8 (2015) 1885-1891. DOI:10.1002/cssc.201403398 |
| [6] |
M.M. Villaverde, N.M. Bertero, T.F. Garetto, A.J. Marchi, Catal. Today 213 (2013) 87-92. DOI:10.1016/j.cattod.2013.02.031 |
| [7] |
C.P. Jiménez-Gómez, J.A. Cecilia, D. Durán-Martín, et al., J. Catal. 336 (2016) 107-115. DOI:10.1016/j.jcat.2016.01.012 |
| [8] |
B.M. Nagaraja, V.S. Kumar, V. Shasikala, et al., Catal. Commun. 4 (2003) 287-293. DOI:10.1016/S1566-7367(03)00060-8 |
| [9] |
M. Ghashghaee, S. Sadjadi, S. Shirvani, V. Farzaneh, Catal. Lett. 147 (2017) 318-327. DOI:10.1007/s10562-016-1948-6 |
| [10] |
C.P. Jimenez-Gomez, J.A. Cecilia, D. Duran-Martin, et al., J. Catal. 336 (2016) 107-115. DOI:10.1016/j.jcat.2016.01.012 |
| [11] |
D. Vargas-Hernandez, J.M. Rubio-Caballero, J. Santamaria-Gonzalez, et al., J. Mol. Catal. A: Chem. 383 (2014) 106-113. |
| [12] |
H.X. Li, H.S. Luo, L. Zhuang, et al., J. Mol. Catal. A: Chem. 203 (2003) 267-275. DOI:10.1016/S1381-1169(03)00368-6 |
| [13] |
X.F. Chen, H.X. Li, H.S. Luo, M.H. Qiao, Appl. Catal. A: Gen. 233 (2002) 13-20. DOI:10.1016/S0926-860X(02)00127-8 |
| [14] |
H.J. Guo, H.R. Zhang, L.Q. Zhang, et al., Ind. Eng. Chem. Res. 57 (2018) 498-511. DOI:10.1021/acs.iecr.7b03699 |
| [15] |
B.M. Reddy, G.K. Reddy, K.N. Rao, et al., J. Mol. Catal. A: Chem. 265 (2007) 276-282. DOI:10.1016/j.molcata.2006.10.034 |
| [16] |
L. Qi, I.T. Horvath, ACS Catal. 2 (2012) 2247-2249. DOI:10.1021/cs300428f |
| [17] |
S. Enthaler, J. von Langermann, T. Schmidt, Energy Environ. 3 (2010) 1207-1217. DOI:10.1039/b907569k |
| [18] |
W.D. Yang, P.L. Li, D.C. Bo, H.Y. Chang, Carbohydr. Res. 357 (2012) 53-61. DOI:10.1016/j.carres.2012.05.020 |
| [19] |
L. He, J. Ni, L.C. Wang, et al., Chemistry 15 (2009) 11833-11836. DOI:10.1002/chem.200901261 |
| [20] |
C.K.P. Neeli, Y.M. Chung, W.S. Ahn, ChemCatChem 9 (2017) 4570-4579. DOI:10.1002/cctc.201701037 |
| [21] |
J. Du, J. Zhang, Y. Sun, et al., J. Catal. 368 (2018) 69-78. DOI:10.1016/j.jcat.2018.09.025 |
| [22] |
G. Muthu Kumaran, S. Garg, K. Soni, et al., Microporous Mesoporous Mater. 114 (2008) 103-109. DOI:10.1016/j.micromeso.2007.12.021 |
| [23] |
J. Xiao, D. Mao, X. Guo, J. Yu, Energ. Technol. 3 (2015) 32-39. DOI:10.1002/ente.201402091 |
| [24] |
Y. Zhu, Y. Zhu, G. Ding, et al., Appl. Catal. A: Gen. 468 (2013) 296-304. DOI:10.1016/j.apcata.2013.09.019 |
| [25] |
M.H. Lim, C.F. Blanford, A. Stein, J. Am. Chem. Soc. 119 (1997) 4090-4091. DOI:10.1021/ja9638824 |
| [26] |
K. Li, Y. Wang, S. Wang, et al., J. Nat. Gas Chem. 18 (2009) 449-452. DOI:10.1016/S1003-9953(08)60144-9 |
| [27] |
Q. Hu, G. Fan, L. Yang, F. Li, ChemCatChem 6 (2014) 3501-3510. DOI:10.1002/cctc.201402674 |
| [28] |
S.P. Wang, X.C. Zheng, X.Y. Wang, et al., Catal. Lett. 105 (2005) 163-168. DOI:10.1007/s10562-005-8686-5 |
| [29] |
S. Srivastava, G.C. Jadeja, J. Parikh, RSC Adv. 6 (2016) 1649-1658. DOI:10.1039/C5RA15048E |
| [30] |
J. Wang, P.A. Chernavskii, A.Y. Khodakov, Y. Wang, J. Catal. 286 (2012) 51-61. DOI:10.1016/j.jcat.2011.10.012 |
| [31] |
Y. Hao, M. Li, F. Cárdenas-Lizana, M.A. Keane, Catal. Struct. React. 1 (2016) 132-139. |
| [32] |
V. Labalme, E. Garbowski, N. Guilhaume, M. Primet, Appl. Catal. A: Gen. 138 (1996) 93-108. DOI:10.1016/0926-860X(95)00248-0 |
| [33] |
C. Qiao, X.F. Liu, X. Liu, L.N. He, Org. Lett. 19 (2017) 1490-1493. DOI:10.1021/acs.orglett.7b00551 |
| [34] |
S. Poulston, E. Rowbotham, P. Stone, et al., Catal. Lett. 52 (1998) 63-67. DOI:10.1023/A:1019007100649 |
| [35] |
R.V. Sharma, U. Das, R. Sammynaiken, A.K. Dalai, Appl. Catal. A: Gen. 454 (2013) 127-136. DOI:10.1016/j.apcata.2012.12.010 |