 2021, Vol. 32
2021, Vol. 32 


Carbon dioxide (CO2) has attracted worldwide attention owing to climate change and global warming, and the reduction of CO2 emission has become one of the most urgent issues [1, 2]. Among various sources of CO2 emission, the burning of fossil fuels used for power generation accounts for a significant portion. In order to lessen CO2 emission, CO2 capture from flue gas has become an important and practical approach [3, 4]. On the other hand, methane (CH4) is regarded as a relatively environmental-friendly energy considering its low CO2 emission, but CH4 is principally generated from natural gas (NG) which frequently contains significant quantities of CO2 impurity [5, 6]. The removal of CO2 from CH4 is necessary because the presence of CO2 not only causes the pipeline corrosion during transportation but also lowers the heat value of NG [7]. As a result, CO2 capture and separation (CCS) process has caught public attention and is under urgent investigation due to industrial demand [8], but the industrial scale CCS using liquid amine chemisorbents bears several drawbacks such as the excessive formation of corrosive and toxic matters, and high energy input for the regeneration of amine solutions [9, 10]. In contrast, physisorption-based separation utilizing porous materials as physisorbents is known to be a promising alternative for CO2 removal [11, 12], but the development of high-performance adsorbents with high CO2 adsorption capacity and efficient separation of CO2/N2 and CO2/CH4 mixtures has become a key factor.
During the past decades, the newly emerged porous metal-organic frameworks (MOFs) have attracted great attention owing to their designable porosity, decoratable framework, moderate CO2 affinity and suitable adsorption kinetics, and great efforts have been devoted to their potential applications in gas storage and separation [13-16]. In order to develop MOFs for efficient CO2 separation, some effective strategies have been applied. The introduction of functional groups (such as -CH3, -NO2, -NH2, -OH, -COOH) into the pores can adjust the polarity and acidity of the porous environment, thus offering higher affinity towards CO2 to boom adsorption capacity and selectivity [17, 18]. For example, Zheng et al. reported a Cd-MOF with phenolic hydroxyl groups as hydrogen-bonding donors displaying better CO2 sorption capacity as compared to the unmodified one [19]. Another approach to facilitate CCS process is to create open metal sites (OMS), which are usually generated by removal of weakly coordinated solvents [20]. Liu et al. reported two Cu-MOFs where the CO2 molecules are apt to locate in the open Cu metal sites via Grand Canonical Monte Carlo (GCMC) simulation [21]. However, removal of coordinated solvents in many instances would cause collapse of the whole framework, or the metal site might transform the coordination geometry into a thermodynamically more stable form instead of forming OMS [22]. Consequently, development of stable porous MOF with OMS remains a great challenge.
Recently, we attempted constructing new MOF utilizing N, N′-bis(4-carboxy-2-methylphenyl)pyromellitic di-imide) as ligand. Fortunately or unfortunately, during the assembly with Ba2+, this methyl-functionalized ligand experienced an unexpected in-situ hydrolysis reaction, and a new barium-based MOF, [H2N(CH3)2]0.5[Ba1.5(L)(DMA)]·1.5DMA·1.5H2O (UPC-70), was obtained based on the partial hydrolysate (2-(4-carboxy-2-methylphenyl)-1, 3-dioxoisoindoline-5, 6-dicarboxylic acid, H3L (Scheme S1 in Supporting information). The crystal structure analysis reveals that UPC-70 contains two different types of 1D open channels with different sizes (24 Å and 10 Å) and abundant OMS after removal of solvents, which provide the possibility of CO2 capture. The gas sorption and separation experiments reveal its high separation selectivity for CO2/CH4 (15) and CO2/N2 (32) at ambient conditions and GCMC simulation further confirms the preferential binding sites of CO2 in UPC-70, indicating its potential as a candidate for selective CO2 separation from CH4 and N2.
The methyl-functionalized ligand was synthesized via one-step condensation of 1, 2, 4, 5-benzenetetracarboxylic anhydride and 4-amino-3-methylbenzoic acid, and the synthetic details were listed in Supporting information. The solvothermal reaction of ligand and Ba(NO3)2 in DMA/Dioxane/H2O (5/2/1, v/v/v) afforded block crystals of UPC-70, and the crystallographic data are summarized in Tables S1-S3 (Supporting information).
Single-crystal X-ray diffraction analysis reveals that UPC-70 crystallizes in the trigonal system with the P-3c1 space group. In the asymmetric unit, there exist one and a half crystallographically unique Ba2+ cations, a deprotonated L3-, a DMA molecule, a H2O molecule and a half free [NH2(CH3)2]+ cation generated from the decomposition of DMA (Fig. 1a). Ba1 is coordinated by six carboxylate oxygen atoms from four ligands, and Ba2 is coordinated by five carboxylate oxygen atoms from four ligands, a coordinated DMA molecule and a coordinated H2O molecule. Adjacent Ba2+ cations are bridged by carboxylates to form catenary secondary building units (SBUs) and each ligand is linked to two chains, accordingly giving rise to a 3D framework with two kinds of 1D channels (Ⅰ: 24 Å, Ⅱ: 10 Å) along the c axis (Figs. 1b- d). It is worth noting that the coordinated DMA molecules and the modified methyl functional groups are directed into the triangular channel Ⅱ, while the coordinated H2O molecules are directed into the hexagonal channel Ⅰ, which is occupied by [NH2(CH3)2]+ cations via hydrogen bonding interactions between [NH2(CH3)2]+ and carboxylate O atoms (C…O 2.838 Å; C-H…O 129.501°, Fig. 1a). PLATON calculation reveals that a total solvent-accessible volume of UPC-70 is 9741.0 Å3 per unit cell, which accounts for approximately 68.4% of the cell volume [23].

|
Download:
|
| Fig. 1. Crystal structure of UPC-70 indicating (a) asymmetric unit; (b) coordination mode of ligand; (c) 1D catenary SBU; (d) 3D packing diagram along the c axis. | |
{kind=link}
The phase purity of UPC-70 was independently confirmed by powder X-ray diffraction (PXRD) patterns (Fig. S2 in Supporting information). The thermogravimetric analysis (TGA) shows that the as-synthesized UPC-70 lost all solvent molecules at about 430 ℃ (Fig. S3 in Supporting information). This is a two-step weight loss corresponding to the guest and coordinated solvent molecules, respectively. The further rising temperature leads to the framework decomposition.
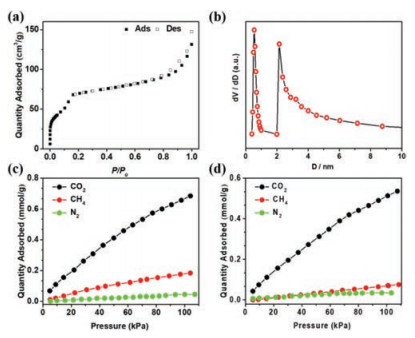
The permanent porosity of UPC-70 is confirmed by N2 adsorption-desorption isotherms at 77 K (Fig. 2a), which show a type-Ⅳ isotherm with the maximum uptake of 147.6 cm3/g. The BET and Langmuir surface areas are estimated to be 239.1 and 421.6 m2/g, and the pore volume is calculated to be 0.23 cm3/g. It should be noted that the pore volume is far lower than the theoretical value of 0.85 cm3/g estimated from the crystal structure, which is typical of the flexible MOF because of the structural contractions during activation [24]. A pore-size distribution is analysed by non-local density functional theory (NLDFT) utilizing N2 adsorption isotherm at 77 K, revealing thatUPC-70 exhibits a narrow pore-size distribution of ~5 and ~20 Å (Fig. 2b).

|
Download:
|
| Fig. 2. (a) N2 adsorption-desorption isotherms at 77 K. (b) Pore-size distribution. CH4, N2 and CO2 sorption isotherms of UPC-70 at (c) 273 K and (d) 298 K. | |
{kind=link}
The micro-pore channels along with the OMS after activation and the existence of [NH2(CH3)2]+ in UPC-70 inspired us to investigate its potential application in gas separation, and CO2 adsorption over CH4 and N2 studies were carried out at 298 and 273 K, respectively (Figs. 2c and d). At 1 atm and 298 K, UPC-70 showed different capacities for CO2 (0.53 mmol/g), CH4 (0.07 mmol/g) and N2 (0.03 mmol/g). At 273 K, the corresponding adsorbed amounts of CO2, CH4, and N2 are 0.68, 0.18 and 0.04 mmol/g, respectively. The adsorption amounts of CO2 at both temperatures and 1 atm are obviously higher than those of CH4 and N2, which can be attributed to the collaborative effect of small pore size, the presence of methyl functional groups and [NH2(CH3)2]+ on the microporous surface [25]. The difference in adsorption performance inspired us to explore its potential in selective separation of CO2/CH4 and CO2/N2. To better understand the gas sorption behavior of UPC-70, the coverage-dependent adsorption enthalpies (Qst) on CO2, CH4, and N2 were calculated by using the virial method. As shown in Fig. S5 (Supporting information), the Qst at near zero-coverage of UPC-70 is 17.7 kJ/mol for CO2. In contrast, the lower Qst of CH4 and N2 suggests weaker adsorbate-adsorbent interactions, which is understandable due to the higher quadrupole moment and polarizability of CO2 compared with that of CH4 and N2 [26].
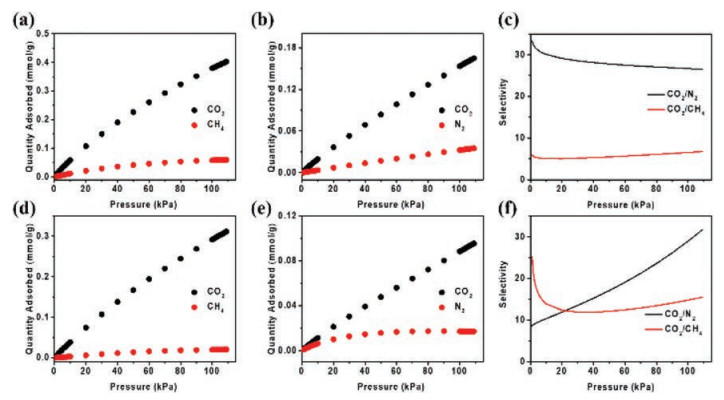
In order to predict gas selectivities of UPC-70, the ideal adsorbed solution theory (IAST) calculation is employed for mixed CO2/CH4 (50%: 50%), and CO2/N2 (15%: 85%) adsorption at different pressures and temperatures [27]. As shown in Fig. 3, the pressure independent isotherm and selectivity profiles indicate relatively good CO2/CH4 and CO2/N2 separation performance. At 1 atm, the selectivity of CO2/CH4 is calculated to be 15 at 298 K and 6 at 273 K, while the selectivity of CO2/N2 is 32 at 298 K and 26 at 273 K. Though the sorption capacity is somewhat small, it is worth to note that the CO2/N2 selectivity surpasses those of some reported MOFs, such as MOF-5 (9) [28], ZIF-300 (22) [29], LIFM-10 (14.5) [30] and SIFSIX-2-Cu (13.7) [31], and the CO2/CH4 selectivity is higher than those of LIFM-10 (4.3) [30], SIFSIX-2-Cu (5) [31], ZJNU-19 (6.4) [32] and ZJU-15 (4) [33], and the corresponding data are summarized in Tables S4 and S5 (Supporting information). Generally, the impressive adsorption selectivity demonstrates that the framework ofUPC-70 can form stronger interactions with CO2 than CH4 or N2, indicating its potential in CO2 capture from flue gas and natural gas.

|
Download:
|
| Fig. 3. Mixture adsorption isotherms at (a, b) 273 K and (d, e) 298 K, and selectivities predicted by IAST calculation for CO2/CH4 (50%: 50%) and CO2/N2 (15%: 85%) separation at (c) 273 K and (f) 298 K. | |
{kind=link}
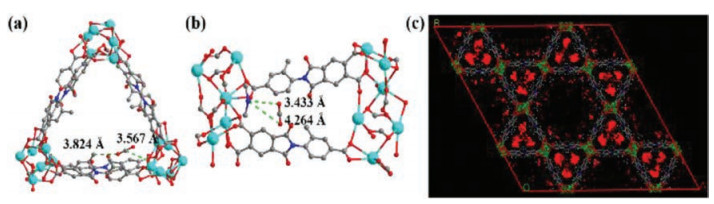
To further investigate interactions between CO2 and UPC-70, GCMC simulations were carried out to explore the most preferential CO2 binding sites. The results reveal that UPC-70 displays most preferential binding sites involving [NH2(CH3)2]+ cations, methyl groups, and carboxylate oxygen atoms. CO2 was trapped in the channels and located around the carboxylate oxygen atoms, methyl groups, and [NH2(CH3)2]+ cations embedded within the pores, with the short distances of C…O (3.824 and 3.567 Å) and N…O (3.433 and 4.264 Å) (Figs. 4a and b). The CO2 adsorbed in the pores of UPC-70 is broadly distributed in channel Ⅱ (Fig. 4c), which can explain the relatively small capacity. The simulation results may suggest that there may exist stronger binding interactions between CO2 and the active sites within the MOFs, which have a synergistic effect on enhancing the CO2 separation selectivity.

|
Download:
|
| Fig. 4. (a, b) GCMC simulation results displaying the preferential CO2 binding sites of UPC-70 and (c) distribution of CO2 adsorbed in UPC-70, viewed from c axis. | |
{kind=link}
In summary, a 3D hierarchical meso-microporous Ba-MOFs with two different types of 1D channels was obtained based on the partial hydrolysate of N, N′-bis(4-carboxy-2-methylphenyl)pyromellitic di-imide), and the selective adsorption studies were carried out. The selective CO2/CH4 and CO2/N2 adsorption can be attribute to the collaborative effect of abundant [NH2(CH3)2]+ cations, unsaturated metal sites, modification of methyl groups and the suitable pore size. The IAST calculation further evaluates the selective separation potential of the binary-component adsorption behavior, making it a potential candidate for CO2 capture from flue gas and natural gas. Moreover, the GCMC simulation reveals that there exist preferential stronger interactions between the active sites and CO2 molecules. This work demonstrates the CO2 selective adsorption performance of UPC-70, providing insight into future design of MOF for CO2/CH4 and CO2/N2 separation.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (NSFC, No. 21771191) and the Fundamental Research Funds for the Central Universities (No. 19CX05001A).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.\09.036.
| [1] |
J. Gong, M. Antonietti, J. Yuan, Angew. Chem. Int. Ed. 56 (2017) 7557-7563. DOI:10.1002/anie.201702453 |
| [2] |
T. Rojek, L. Gubler, M.M. Nasef, E. Abouzari-Lotf, Ind. Eng. Chem. Res. 56 (2017) 5925-5934. DOI:10.1021/acs.iecr.7b00862 |
| [3] |
E.S. Sanz-Pérez, C.R. Murdock, S.A. Didas, C.W. Jones, Chem. Rev. 116 (2016) 11840-11876. DOI:10.1021/acs.chemrev.6b00173 |
| [4] |
R. Lyndon, K. Konstas, B.P. Ladewig, et al., Angew. Chem. Int. Ed 52 (2013) 3695-3698. DOI:10.1002/anie.201206359 |
| [5] |
Q. Zhang, J. Wang, T. Wang, Ind. Eng. Chem. Res. 56 (2017) 5174-5184. DOI:10.1021/acs.iecr.7b00406 |
| [6] |
K.J. Chen, D.G. Madden, T. Pham, et al., Angew. Chem. Int. Ed 55 (2016) 10268-10272. DOI:10.1002/anie.201603934 |
| [7] |
S. Feng, Y. Li, H. Liu, et al., J. Nat. Gas Sci. Eng. 80 (2020) 103395. DOI:10.1016/j.jngse.2020.103395 |
| [8] |
R. Ben-Mansour, M.A. Habib, O.E. Bamidele, et al., Appl. Energy 161 (2016) 225-255. DOI:10.1016/j.apenergy.2015.10.011 |
| [9] |
N. Tippayawong, P. Thanompongchart, Energy 35 (2010) 4531-4535. DOI:10.1016/j.energy.2010.04.014 |
| [10] |
Z.J. Zhang, Z.Z. Yao, S.C. Xiang, B.L. Chen, Energy Environ. Sci. 7 (2014) 2868-2899. DOI:10.1039/C4EE00143E |
| [11] |
Y. Liu, Z.U. Wang, H.C. Zhou, Greenhouse Gases: Sci. Technol. 2 (2012) 239-259. DOI:10.1002/ghg.1296 |
| [12] |
M. Khraisheh, F. Almomani, G. Walker, Sci. Rep. 10 (2020) 269. DOI:10.1038/s41598-019-57151-x |
| [13] |
W.G. Cui, G.Y. Zhang, T.L. Hu, X.H. Bu, Coord. Chem. Rev. 387 (2019) 79-120. DOI:10.1016/j.ccr.2019.02.001 |
| [14] |
Y. Ye, R.B. Lin, H. Cui, et al., Dalton Trans. 49 (2020) 3658-3661. DOI:10.1039/C9DT01911A |
| [15] |
L. Chen, R. Luque, Y. Li, Chem. Soc. Rev. 46 (2017) 4614-4630. DOI:10.1039/C6CS00537C |
| [16] |
Q. Gao, J. Xu, D. Cao, Z. Chang, X.H. Bu, Angew. Chem. Int. Ed 55 (2016) 15027-15030. DOI:10.1002/anie.201608250 |
| [17] |
P. Cui, Y.G. Ma, H.H. Li, et al., J. Am. Chem. Soc. 134 (2012) 18892-18895. DOI:10.1021/ja3063138 |
| [18] |
M.H. Yu, B. Space, D. Franz, et al., J. Am. Chem. Soc. 141 (2019) 17703-17712. DOI:10.1021/jacs.9b07807 |
| [19] |
Z.J. Wang, L.J. Han, X.J. Gao, H.G. Zheng, Inorg. Chem. 57 (2018) 5232-5239. DOI:10.1021/acs.inorgchem.8b00272 |
| [20] |
Y. Wang, M. He, X. Gao, S. Li, Y. He, Dalton Trans. 47 (2018) 7213-7221. DOI:10.1039/C8DT00863A |
| [21] |
B. Liu, S. Yao, X. Liu, et al., ACS Appl. Mater. Interfaces 9 (2017) 32820-32828. DOI:10.1021/acsami.7b10795 |
| [22] |
C. Atzori, K.A. Lomachenko, S. Øien-Ødegaard, et al., Cryst. Growth Des. 19 (2019) 787-796. DOI:10.1021/acs.cgd.8b01369 |
| [23] |
A.L. Spek, Acta Crystallogr. Sect. C: Struct. Chem. 71 (2015) 9-18. DOI:10.1107/S2053229614024929 |
| [24] |
M.Y. Kan, J.H. Shin, C.T. Yang, et al., Chem. Mater. 31 (2019) 7666-7677. DOI:10.1021/acs.chemmater.9b02539 |
| [25] |
H.Y. Ma, Y.Z. Zhang, H. Yan, et al., Dalton Trans. 48 (2019) 13541-13545. DOI:10.1039/C9DT02694K |
| [26] |
Y. Chen, D. Lv, J. Wu, et al., Chem. Eng. J. 308 (2017) 1065-1072. DOI:10.1016/j.cej.2016.09.138 |
| [27] |
K.S. Walton, D.S. Sholl, AIChE J. 61 (2015) 2757-2762. DOI:10.1002/aic.14878 |
| [28] |
N. Ding, H. Li, X. Feng, et al., J. Am. Chem. Soc. 138 (2016) 10100-10103. DOI:10.1021/jacs.6b06051 |
| [29] |
N.T.T. Nguyen, H. Furukawa, F. Ga'ndara, et al., Angew. Chem. Int. Ed. 53 (2014) 10645-10648. DOI:10.1002/anie.201403980 |
| [30] |
Y. Xiong, Y.Z. Fan, R. Yang, et al., Chem. Commun. 50 (2014) 14631-14634. DOI:10.1039/C4CC06697A |
| [31] |
P. Nugent, Y. Belmabkhout, S.D. Burd, et al., Nature 495 (2013) 80-84. DOI:10.1038/nature11893 |
| [32] |
T. Xu, L. Fan, Z. Jiang, et al., Dalton Trans. 49 (2020) 7174-7181. DOI:10.1039/D0DT01081B |
| [33] |
X. Duan, Y. Zhou, R. Lv, et al., J. Solid State Chem. 260 (2018) 31-33. DOI:10.1016/j.jssc.2018.01.002 |