 2021, Vol. 32
2021, Vol. 32 


b CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology (NCNST), Beijing 100190, China;
c Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China;
d Laboratory of Theoretical and Computational Nanoscience, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China;
e Center of Materials Science and Optoelectonics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China;
f Sino-Danish Center for Education and Research, University of Chinese Academy of Sciences, Beijing 100190, China
Liquid crystals are basically dynamic functional materials that combine order and mobility at molecular and macroscopic levels [1]. Since the discovery by Reinitzer in 1888 [2], liquid crystals have been studied in multiple disciplines as prototype self-assembling supramolecular soft materials, because of their involvement of almost all kinds of interactions including van der Walls interaction, hydrogen bonding interaction, dipolar interaction, metal coordination and so on [1, 3]. Other than the well-known technological applications in the liquid-crystal display (LCD) devices [4-6], liquid crystals can also be used as functional materials for electron and ion transporting [7, 8], sensing [9, 10], catalysis [11, 12], templates [13-15] and biomedicine [16, 17], etc.
Self-assembly is a spontaneous organization of components into regular patterns or structures, which has been extensively investigated for decades with the main goal of providing access to functional materials [18-22]. During the molecular organization process, the supramolecular structure is primarily determined by the intermolecular interactions, which correlate with the functional groups to a great extent. Hence the effect of functional groups on self-assembly has drawn more and more attention. Valera et al. [23] investigated the supramolecular assembly of two pyrene derivatives, governed by the C–H...π interactions and the H-bonding interaction between the amide groups and the imidazole moieties. The pyrene-based aggregate preserves the blue-emitting properties of the monomer, showing the potential application in Organic Electronics. Aparicio et al. [24] synthesized three cyano-substituted divinylene arene-based luminogens as fluorescent π-gelators. The donor-acceptor interaction in self-assembly can be modulated by changing the position of cyano groups in the conjugated backbone, thus achieving the modulation of the emission color. Recently, Rödle et al. [25] have also compared the supramolecular polymerization of two liquid crystal molecules that only differ in the linking group to investigate the influence of amide and ester groups on the molecular arrangement. They revealed the rotationally displaced organization for the amide‐containing molecules and the parallel arrangement for the ester-containing molecules. However, by using spectroscopic methods, the assembly structures of aggregates were too complicated to be characterized directly and clearly enough.
Therefore, to further investigate the impact of functional groups on the supramolecular assembly mechanism, herein we report the explicit self-assembly structures of three liquid crystal molecules which are similar in molecular structure. As shown in Scheme 1, we employed three liquid crystal molecules which only differ in the functional groups of backbones as the research objects. The difference between LC-amide-1 and LC-ester is the amide/ester groups at both ends of the backbone, while LC-amide-1 and LC-amide-2 differ in the presence/absence of central BODIPY group. The LC-amide-1, LC-amide-2 and LC-ester are separately dissolved in 1-phenyloctane, wherein the concentrations are less than 10 −3 mol/L. Then the assembly samples were prepared by depositing a droplet of the corresponding solution (0.1 μL) onto the freshly cleaved highly oriented pyrolytic graphite (HOPG) surface, respectively. Afterwards, the uniform and regular assembly structures were precisely revealed at the HOPG/1-phenyloctane interface by using STM under ambient conditions. The details of sample preparation and STM characterization procedures can be found in Supporting Information. In combination with DFT calculation, the self-assembly mechanism was deeply explored.

|
Download:
|
| Scheme1. Chemical structures of (a) LC-amide-1, (b) LC-ester and (c) LC-amide-2. | |
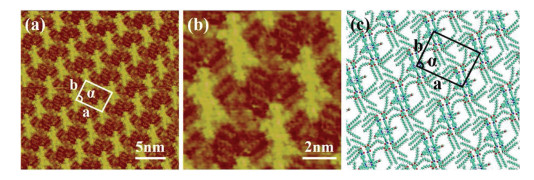
First, the assembly structure of LC-amide-1 at the HOPG/1-phenyloctane interface is characterized by using STM. Fig. 1a presents the large scale STM image of the assembly structure, in which the bright rods are aligned in parallel and form the uniform linear pattern. The average length of bright rods is measured to be 4.0 ± 0.1 nm, which shows good agreement with the theoretical size of the backbone of LC-amide-1 molecules. And the alkyl chains of LC-amide-1 molecules correspond to the lines distributed in the shade, which cannot be explicitly characterized due to the much lower density of electric states. Further structural details are displayed in high resolution STM image, as shown inFig. 1b, indicating that each rod element is a dimer formed by two closed-packed LC-amide-1 molecules. The two molecules in the dimer can interact with each other through N-H···O hydrogen bonding between amide groups. Besides, the simulated molecular packing structure as shown inFig. 1c indicates that the benzene rings at both ends of the molecule are almost perpendicular to the corresponding benzene rings of another molecule in the dimer, which can form π-π stacking interaction. It can also be distinguished that the alkyl chains are distributed in a staggered form and stretched in different directions. The van der Waals interaction between these alkyl chains, the π-π stacking interaction between corresponding benzene rings and the π-π stacking interaction between two BODIPY moieties in the dimer are the primary assembly motifs of the linear structure. The parameters of the unit cells overlaid in Fig. 1a are measured as follows: a = 4.8 ± 0.1 nm, b = 3.5 ± 0.1 nm, α = 85 ± 1°.

|
Download:
|
| Fig. 1. STM images of LC-amide-1 assembly structure at the HOPG/1-phenyloctane interface: (a) Large scale; (b) high resolution. Tunneling conditions: Iset = 219.7 pA, Vbias =734.3 mV. (c) The simulated molecular packing structure. | |
In the next step, the self-assembly of LC-ester at the HOPG/1-phenyloctane interface is investigated. Fig. 2a presents the large scale STM image of LC-ester assembly structure. It can be observed that the butterfly-shaped dimers are arranged in parallel into the linear pattern. And the orientation of dimers is at an angle to the direction of the rows. As shown in the high resolution STM image inFig. 2b, each dimer consists of two parallel bright rods. The average length of bright rods is measured to be 3.8±0.1 nm, which is consistent with the theoretical size of the backbone of LC-ester. Therefore, it can be concluded that the butterfly-shaped dimers are composed of two opposite LC-ester molecules, which mostly interact through π-π stacking interaction between BODIPY moieties and benzene rings. Combined with the simulated molecular packing structure in Fig. 2c, it can be concluded that the parallel lines distributed between the neighbor rows are attributed to the alkyl chains of LC-ester molecules. The assembly motif derives from both intermolecular π−π stacking interaction and the van der Waals interaction between alkyl chains. The parameters of the unit cells overlaid in Fig. 2b are measured as follows: a = 4.3 ± 0.1 nm, b = 4.3 ± 0.1 nm, α = 105 ± 1°.

|
Download:
|
| Fig. 2. STM images of LC-ester assembly structure at the HOPG/1-phenyloctane interface: (a) Large scale; (b) high resolution. Tunneling conditions: Iset = 228.9 pA, Vbias =714.7 mV. (c) The simulated molecular packing structure. | |
LC-amide-2 molecules can also form regular self-assembly structure at the HOPG/1-phenyloctane interface. Fig. 3a presents the large scale STM image of LC-amide-2 assembly structure, in which the bright rods aligned in parallel can be observed. As shown in the high resolution STM image in Fig. 3b, the rods containing two bright spots at both ends are measured to be 2.7 ± 0.1 nm, which shows good agreement with the theoretical size of the backbone of LC-amide-2 molecules. Therefore, it can be inferred that the bright rods correspond to the LC-amide-2 molecules and the bright spots correspond to the benzene rings at both ends of the backbone. The acetylene linkages and alkyl chains cannot be explicitly characterized due to the much lower density of electric states. Combined with the simulated molecular packing structures shown in Fig. 3c, it can be inferred that the alkyl chains are aligned into one direction and the amide groups are too far apart to form hydrogen bonds. Therefore, the assembly motif mostly derives from the van der Waals interaction between these alkyl chains. The parameters of the unit cells overlaid in Fig. 3b are measured as follows: a = 3 ± 0.1 nm, b = 2.5 ± 0.1 nm, α = 110 ± 2°.

|
Download:
|
| Fig. 3. STM images of LC-amide-2 assembly structure at the HOPG/1-phenyloctane interface: (a) Large scale; (b) high resolution. Tunneling conditions: Iset = 222.7 pA, Vbias =693.2 mV. (c) The simulated molecular packing structure. | |
To better understand the self-assembly structures of three liquid crystal molecules, the unit cell parameters and interaction energies are calculated by the DFT method based on the STM characterizations. The details of DFT calculation can be found in the Supporting Information. Table 1 lists the calculated unit cell parameters of three assembly systems, which agree well with the corresponding experimental results. The interaction energies of three self-assemblies are presented inTable 2 and the lower energy indicates the stronger interaction herein. It can be observed that the interaction energy between LC-amide-1 molecules (-149.536 kcal/mol) is lower than that between LC-ester molecules (-134.914 kcal/mol), which is largely due to the extra N-H···O hydrogen bonds between amide groups of LC-amide-1 molecules. And the interaction energy between LC-amide-2 molecules (-16.254 kcal/mol) is much higher because LC-amide-2 molecules mainly interact through the weak van der Waals interaction. Besides the interaction between assembled molecules, the interaction between molecules and substrate also plays a crucial part in the surface assembly. As shown in the second column inTable 2, the interaction energy between LC-amide-1 molecules and substrate (-418.567 kcal/mol) is similar to that between LC-ester molecules and substrate (-421.721 kcal/mol), which are both lower than that between LC-amide-2 molecules and substrate (-152.124 kcal/mol) due to the π-π stacking interaction between BODIPY groups and graphite substrate. The interaction energies between molecules and substrate of three assembly systems are all much lower than the interaction energies between molecules, indicating that the absorption between molecules and HOPG substrate is quite strong.
|
|
Table 1 Experimental (Expt.) and calculated (Cal.) unit cell parameters of the self-assemblies on the HOPG surface. |

|
|
Table 2 Total energies and energies per unit area of self-assemblies on the HOPG surfacea. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The total energies (including the interaction energies between molecules and the interaction energies between molecules and substrate) of three self-assembly systems are presented in Table 2. In general, the total energy can be compared to evaluate the relative thermodynamic stability of different assembly systems with the same unit cell. However, the effect of the unit area should be considered when comparing two systems with different unit cells. For the assembly system with the smaller unit cell, more molecules would be adsorbed on the surface within the same area, thus contributing more interaction energy to the system.[26] Therefore, the total energy per unit area is also calculated to avoid such effect. As displayed in the last column in Table 2, the total energy per unit area of LC-amide-2 system (-0.234 kcalmol−1 Å -2) is the highest and the total energy per unit area of LC-amide-1 system (-0.338 kcalmol−1 Å-2) is slightly lower than that of the LC-ester system (-0.303 kcalmol−1 Å -2), which suggests that the self-assembly of LC-amide-1 is most thermodynamically stable.
As mentioned above, the three liquid crystal molecules all assemble into the linear patterns at the HOPG/1-phenyloctane interface. However, the differences in the backbone of molecules lead to different linear structures. As for the self-assembly of LC-amide-1 and LC-ester, the linear patterns are both formed by the arrangement of dimers. The two liquid crystal molecules in the dimer can interact through π-π stacking interaction. Moreover, the amide moieties in LC-amide-1 molecules can form hydrogen bonds, while the ester moieties in LC-ester molecules cannot. Therefore, it can be distinguished that the two LC-amide-1 molecules in the dimer are arranged much closer comparing to the arrangement of LC-ester dimer, which agrees well with the result that the interaction energy between LC-amide-1 molecules (-149.536 kcal/mol) is lower than that between LC-ester molecules (-134.914 kcal/mol). As for the self-assembly of LC-amide-2, the linear pattern is formed by the arrangement of molecules instead of dimers. The van der Waals interaction between alkyl chains is the primary assembly motif, and the alkyl chains are arranged into one direction to maximize the van der Waals interaction. Moreover, as shown inFig. 3, the lowermost end of the backbone of the LC-amide-2 molecule is aligned with the uppermost end of the backbone of the lower right molecule to make the alkyl chains be aligned and interact more with each other. Hence the amide groups are too far apart to form the hydrogen bonds. Considering that the only difference between the molecular structures of LC-amide-1 and LC-amide-2 is the BODIPY group, it can be inferred that the π-π stacking interaction is also important in forming dimers. Combined with the DFT result that the interaction energy between LC-amide-1 molecules (-149.536 kcal/mol) is much lower than that of LC-amide-2 molecules (-16.254 kcal/mol), it can be inferred that the interaction between LC-amide-2 molecules is not strong enough to form dimers with the absence of π-π stacking interaction and hydrogen bonding interaction. Considering the experimental results and DFT calculations of three self-assembly systems, it is indicated that the simultaneous presence of N-H···O hydrogen bonding between amide groups, van der Waals interaction between alkyl chains, π-π stacking interaction between BODIPY groups and between benzene rings results in the closest molecular arrangement and the best thermodynamic stability of LC-amide-1 system in three liquid crystal self-assemblies.
To sum up, by comparing the assembly structures of three liquid crystal molecules that differ in the functional groups at the solid/liquid interface, we have shed light on the influence of these groups on the self-assembly mechanism. The three liquid crystal molecules all form the linear assembly structures and the assembly motifs all include the van der Waals interaction between alkyl chains. However, the other interactions including hydrogen bonding interaction and π-π stacking interaction between molecules containing different functional groups are quite different, leading to the different arrangements of molecules in the linear patterns. The results of the DFT calculation also demonstrate the influence of these functional groups on the self-assembly. This work on the self-assembly of the liquid crystal molecules could enhance our understanding of the supramolecular assembly mechanism and benefit the exploration of the further application of liquid crystals.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 51875303, 21773041, 21972031), the National Basic Research Program of China (No. 2016YFA0200700) and the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDB36000000).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.016.
| [1] |
H.K. Bisoyi, S. Kumar, Chem. Soc. Rev. 40 (2011) 306-319. DOI:10.1039/B901793N |
| [2] |
F. Reinitzer, Monatshefte für Chemie. 9 (1888) 421-441. DOI:10.1007/BF01516710 |
| [3] |
T. Kato, J. Uchida, T. Ichikawa, T. Sakamoto, Angew. Chem. Int. Ed. 57 (2018) 4355-4371. DOI:10.1002/anie.201711163 |
| [4] |
P.S. Drzaic, J. Appl. Phys. 60 (1986) 2142-2148. DOI:10.1063/1.337167 |
| [5] |
M. Schadt, H. Seiberle, A. Schuster, Nature 381 (1996) 212-215. DOI:10.1038/381212a0 |
| [6] |
W.L. He, T. Liu, Z. Yang, et al., Chin. Chem. Lett. 20 (2009) 1303-1306. DOI:10.1016/j.cclet.2009.05.010 |
| [7] |
T. Kato, M. Yoshio, T. Ichikawa, et al., Nat. Rev. Mater. 2 (2017). DOI:10.1038/natrevmats.2017.1 |
| [8] |
B.X. Dong, Z. Liu, M. Misra, et al., ACS Nano 13 (2019) 7665-7675. DOI:10.1021/acsnano.9b01055 |
| [9] |
X. Bi, D. Hartono, K. Yang, Adv. Funct. Mater. 19 (2009) 3760-3765. DOI:10.1002/adfm.200900823 |
| [10] |
S. Yi, H. Gao, Q. Li, et al., Chin. Chem. Lett. 26 (2015) 872-876. DOI:10.1016/j.cclet.2015.05.031 |
| [11] |
Y.J. Xu, W.Q. Gu, D.L. Gin, J. Am. Chem. Soc. 126 (2004) 1616-1617. DOI:10.1021/ja038501i |
| [12] |
D.L. Gin, X.Y. Lu, P.R. Nemade, et al., Adv. Funct. Mater. 16 (2006) 865-878. DOI:10.1002/adfm.200500280 |
| [13] |
C.T. Kresge, M.E. Leonowicz, W.J. Roth, J.C. Vartuli, J.S. Beck, Nature 359 (1992) 710-712. DOI:10.1038/359710a0 |
| [14] |
G.S. Attard, C.G. Goltner, J.M. Corker, S. Henke, R.H. Templer, Angew. Chem. Int. Ed. 36 (1997) 1315-1317. DOI:10.1002/anie.199713151 |
| [15] |
X. Wang, D.S. Miller, E. Bukusoglu, Pablo J.J. de, N.L. Abbott, Nat. Mater. 15 (2016) 106. DOI:10.1038/nmat4421 |
| [16] |
S.Y. Lee, K.I. Park, C. Huh, et al., Nano Energy 1 (2012) 145-151. DOI:10.1016/j.nanoen.2011.07.001 |
| [17] |
Jalili-Firoozinezhad S., M.H.M. Moghadam, M.H. Ghanian, et al., RSC Adv. 7 (2017) 39628-39634. DOI:10.1039/C7RA06178A |
| [18] |
G.M. Whitesides, B. Grzybowski, Science 295 (2002) 2418-2421. DOI:10.1126/science.1070821 |
| [19] |
J.V. Barth, G. Costantini, K. Kern, Nature 437 (2005) 671-679. DOI:10.1038/nature04166 |
| [20] |
H.Y. Shi, X.C. Lu, Y.H. Liu, et al., ACS Nano 12 (2018) 8781-8790. DOI:10.1021/acsnano.8b05045 |
| [21] |
J. Li, X. Zu, Y. Qian, et al., Chin. Chem. Lett. 31 (2020) 10-18. DOI:10.1016/j.cclet.2019.04.032 |
| [22] |
Q. Xue, Y. Zhang, R. Li, et al., Chin. Chem. Lett. 30 (2019) 2355-2358. DOI:10.1016/j.cclet.2019.09.027 |
| [23] |
J.S. Valera, J. Calbo, R. Gómez, E. Ortí, L. Sánchez, Chem. Commun. (Camb.) 51 (2015) 10142-10145. DOI:10.1039/C5CC03616J |
| [24] |
F. Aparicio, S. Cherumukkil, A. Ajayaghosh, L. Sánchez, Langmuir 32 (2016) 284-289. DOI:10.1021/acs.langmuir.5b03771 |
| [25] |
A. Rödle, B. Ritschel, Mück-Lichtenfeld C., V. Stepanenko, G. Fernández, Chem. Eur. J. 22 (2016) 15772-15777. DOI:10.1002/chem.201602592 |
| [26] |
S.Q. Chang, R.C. Liu, L.C. Wang, et al., ACS Nano 10 (2016) 342-348. DOI:10.1021/acsnano.5b06666 |