 2021, Vol. 32
2021, Vol. 32 


Metal oxides are important functional materials and they are widely used in various applications such as catalysis, batteries, optical devices etc., mainly due to their relatively low costs, good stabilities, high activities and favorable redox properties [1]. As one of the most common rare earth metal oxides, cerium dioxide (CeO2) can work as the key component of the catalyst in vehicle emission control, water-gas shift reactions, solid oxide fuel cells and steam reforming [2]. Because of its unique electronic structure, as well as the existence of various types of defects, CeO2 is also often taken as a model material for experimental and theoretical studies in surface chemistry and heterogeneous catalysis [3].
CO oxidation is an important process in the control of vehicle emission and many other catalytic reactions. In particular, CeO2 based catalysts have been found to be very active in promoting CO oxidation, which is also a classical process to illustrate the activity and the oxygen storage capacity (OSC) performance of such catalysts. The highly active surface lattice oxygen (Os) species at CeO2 is determined to play a crucial role in this process, as it generally obeys the Mars-van Krevelen (MvK) [4] mechanism [5, 6]. The whole catalytic cycle follows the following processes:

|
where * stands for a surface oxygen vacancy. Beyond that, the surface lattice oxygen was also found to be able to participate in CO oxidation on the supported metal clusters by directly interacting with adsorbed CO at the metal/CeO2 interface or spilling over to the metal clusters [7]. Therefore, such high activity also leads to the facile removal of Os and formation of reduced CeO2 with Ce3+. During the past few decades, many studies have revealed the relationship between the CO oxidation activity and the concentration of Ce3+ and illustrated that the CeO2 catalyst with a higher Ce3+ concentration usually exhibits a better CO oxidation performance [8]. Some work has concluded this for the high oxygen migration rate provided by oxygen vacancies. However, the detailed connection between the catalytic activity for CO oxidation and the reduction degree of the catalyst is still not clear. Moreover, recent studies suggested that the surface hydroxyls formed by dissociated water at the oxygen vacancy can enhance the activity of CO oxidation, which may also contribute to the high activity of reduced CeO2 catalysts [9]. In fact, besides the findings of the improvement of catalytic activity by surface hydroxyls [10], it is also expected that surface oxygen vacancies can be readily healed by oxygen molecules [11] and therefore they may not be the key species for the enhanced activities in reduced ceria. Accordingly, how the surface Ce3+ itself can affect the catalytic activities of reduced CeO2 toward CO oxidation is still worth studying.
In this work, we conducted density functional theory calculations corrected by on-site Coulomb interaction (DFT+U) to theoretically investigate the effect of the concentrations of Ce3+ [12], which were induced by surface hydroxyls (H at Os) on the catalytic activities toward CO oxidation at CeO2(111), the main facet exposed at CeO2 nano-catalysts. In particular, we mainly focused on the process of direct reaction between CO and surface oxygens to reveal their reactivities at the stoichiometric and various reduced surfaces, though the interaction between adsorbed H or OH and CO on the CeO2 surfaces may be also important [13]. All the calculation details can be found in the Supporting information.
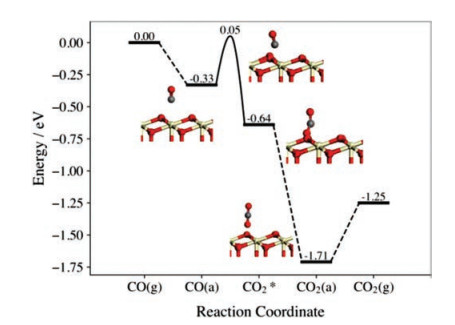
On the pristine CeO2(111) surface, CO can be exothermically adsorbed with the calculated adsorption energy of 0.33 eV (Fig. 1). Then, the CO can react with the Os through the transition state (TS), in which the distance between C and Os (dC-Os) was calculated to decrease from 2.835Å in the adsorption state to 1.665Å (TS) and the ∠OCOs was determined to be 115.81°. The stretching vibration between C and Os in the transition state along this reaction pathway was determined with the imaginary frequency of 268.76 cm−1. The carbon atom in CO would bind with the Os afterwards to form an adsorbed bent CO2* intermediate species with the activation energy of 0.38 eV and the reaction energy of −0.31 eV. The bent CO2* is not stable and prefers to evolve to the straight one by releasing the energy as large as 1.07 eV. The straight CO2 molecule can be easily released from the surface with the desorption energy of 0.46 eV only. These results are largely consistent with those reported in previous theoretical studies of CO oxidation at ceria surfaces [14].

|
Download:
|
| Fig. 1. Calculated energy profile of CO oxidation on the pristine CeO2(111) surface. CO(g) and CO2(g) stand for the CO and CO2 in gas phase, respectively. CO(a) and CO2(a) refer to the adsorbed CO and CO2. CO2* denotes the bent CO2 intermediate. The ivory, red and grey spheres represent the Ce, O and C atoms, respectively. | |
In general, corresponding to the different reaction steps discussed in the above, four important energetic components are involved in the whole CO oxidation process, namely the adsorption energy of CO (Eads), the activation energy (Ea), the reaction energy (Er) and the bending energy (Eb), as shown in Scheme S1 (Supporting information).
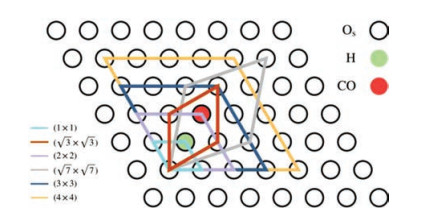
The reduced CeO2(111) surfaces with different concentrations of Ce3+ were constructed by adjusting the coverages of surface hydroxyls (adsorbed hydrogens). A series of surface cells with different sizes involving one adsorbed H were applied as shown in Scheme 1 and Fig. S1 (Supporting information). Accordingly, the coverages (θ) of the adsorbed H are 1/16, 1/9, 1/7, 1/4, 1/3 and 1monolayer (ML) (with respect to the number of Os) depending on the sizes of surface cells. The calculated H adsorption energies on the CeO2(111) surface under the above different coverages are listed in Table 1. One can see that under low coverages (θ ≤ 1/3 ML), the average H adsorption energy is 1.46 eV, while under the highest coverage of 1 ML, the calculated adsorption energy is lower by ~0.1 eV. According to the spin charge difference analysis, the whole injected electron from the adsorbed H is localized at the nearest Ce beside the hydroxyl, which suggests that the varying coverages of adsorbed H can indeed modify the concentrations of Ce3+ on the surface. The occupation of the Ce 4f orbital was also confirmed by the calculated density of states (DOS) (Fig. S2 in Supporting information). It should be noticed that the adsorbed H is introduced to adjust the concentration of Ce3+, which will not directly participate in CO oxidation reaction.

|
Download:
|
| 1. Sketch of the different surface cells and relative positions of the different surface species on the CeO2(111) surface. The black circles filled with green and red stand for the hydroxyl and the reactive Os, respectively. | |
|
|
Table 1 Calculated adsorption energies of hydrogen (Eads(H), with respect to 1/2 H2) on the CeO2(111) surfaces with different surface cells, calculated corresponding average negative charges of Os (Δq(Os) and the band gap (Egap). |

{kind=link}
{kind=link}
Then, at the surfaces with the hydroxyls under the coverages from 1/16 ML to 1/3 ML, we calculated the reaction between one CO and the neighboring Os of the hydroxyl (Scheme 1), and the various energetic components involved in the whole process of the reaction are plotted in Fig. 2 and listed in Table S1 (all corresponding structures and the detailed results of the transition states can be found in Figs. S3-S7 and Table S2 in Supporting information). One can see that the Eads decreases with the increasing Ce3+ concentration. The adsorption energy of CO is 0.35 eV at θ=1/16 ML, which is slightly higher than that on the pristine surface, while it becomes 0.29 eV when the θ increases to 1/3 ML. The calculated Bader charges of adsorbed CO (ΔqIS(CO)) showed that the CO molecule accepts more negative charges at the surface with the increasing reduction degree (Table S3 in Supporting information). In other words, higher concentrations of localized electrons (Ce3+) at CeO2(111) can make the adsorbed CO more negatively charged, though its adsorption strength becomes slightly worse.

|
Download:
|
| Fig. 2. Calculated energetic components (Eads, Ea, Er and Eb) within CO oxidation as a function of the hydroxyl coverages. The detailed results and structures can be found in Table S1 and Figs. S3-S7. | |
{kind=link}
Interestingly, both Ea and Er of CO oxidation apparently decrease with the coverages of surface hydroxyl (Fig. 2). In particular, the calculated Ea drops from 0.43 eV at θ=1/16 ML to 0.31 eV at θ=1/3 ML, and the change of the calculated Er also indicates that more heat can be released through combination of CO and Os on the surface with higher reduction degree. At the same time, in consistence with what was reported in our previous study [6], the Bader charge analysis showed that (ΔqIMS(CO2) in Table S3) the CO2* intermediate formed directly after the combination of CO and Os is actually a negatively charged CO2− species. In this process, the electron-rich Os prefers to attack the partially positively charged C in the CO molecule, and the electron transferred from surface to the CO molecule can be also found from the Bader charge analysis (ΔqTS(CO) – ΔqIS(CO)) in Table S3). Then, one may expect that the localized electrons at Ce3+ on the reduced surface slab can effectively increase the amount of negative charge of the surface Ce layer, which will then affect the charge distribution within the Ce-Os bonds and push the charges toward Os to increase their Δq(Os). Accordingly, one can indeed see from Table 1 that more negatively charged Os can occur at the surface with a higher concentration of surface hydroxyl, which will surely make it more active to be involved in CO oxidation. The activation energy as a function of the Δq(Os) was plotted in Fig. S8 (Supporting information), and one can clearly see that there is a good linear relationship with the R2 of 0.97. The corresponding calculated imaginary frequencies are shown in Table S4 (Supporting information).
The evolvement of the bent CO2− intermediate to a straight CO2 molecule then occurs following the combination of CO and Os, giving rise to the final formation and desorption of the molecular CO2. In this process, the CO2− species leaves one electron to the surface, and it can be confirmed by the calculated charge difference between the FS and IMS (ΔqFS(CO2) – ΔqIMS(CO2)) in Table S3. Accordingly, one may expect that on the reduced CeO2(111) surfaces, the released electron from the CO2− intermediate leads to the occurrence of one more Ce3+, and it is obvious that the energy cost by the occupation of the empty 4f orbitals of Ce4+ by the released electron would affect the bending energy. According to our calculated results, the band gap increases with the increasing concentrations of Ce3+ (Table 1), which indeed suggests that a larger energy is needed to accept the released electron from the CO2− intermediate for the surface with a higher coverage of hydroxyls. Therefore, the corresponding total released energy, Eb, becomes lower. Our previous study [6] suggested that the competitive pathway to form the surface carbonate species rather than the gas phase CO2 may occur at CeO2 due to the high stability of the carbonate, which means that the less Eb would result in the favorable formation of carbonate species. Even though highly reduced ceria catalyst brings a high activity for the combination between CO and Os, the corresponding low selectivity to generate CO2 caused by the Eb would like to cause undesirable effects to the catalyst.
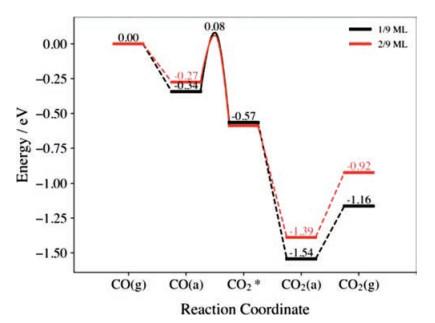
To further verify the effect of surface reduction degree on the catalytic activity, CO oxidation was also calculated on the CeO2(111)-(3×3) surface involving two adsorbed hydrogens (Fig. S9 in Supporting information), and the results and structures are reported in Fig. 3, Fig. S10 and Table S1 (Supporting information). Compared with the CeO2(111)-(3×3) surface involving only one adsorbed H (Fig. S11 in Supporting information), it is then more heavily reduced as two Ce3+ now occur in each surface cell. As one can see, the calculated CO adsorption energy slightly reduces to 0.27 eV and the activation energy also reduces to 0.34 eV, which is 0.08 eV lower than that on the surface with one H (θ=1/9 ML). In addition, the corresponding reaction energy was found to be 0.09 eV lower and the calculated bending energy is 0.18 eV less than those on the surface with only one hydroxyl. These effects can be also related to the surface electronic structures. According to our calculations, the average negative charges of Os on the CeO2(111)-(3×3) surface with two H (θ=2/9 ML) is 1.230 e−. Interestingly, this value is located between those on the surface at θ=1/4 and 1/7 ML (Table 1) and the corresponding activation energy (0.34 eV) is also between those at these two surfaces (Table S1), in agreement with the relationship between the Δq(Os) and the activation energy mentioned above. Therefore, one can indeed see that the surface reduction degree can tune the concentrations of localized electrons at the surface Ce, which in turn modify the charge distributions at the Os and affect their negative charges and activity toward reaction with CO.

|
Download:
|
| Fig. 3. Calculated energy profiles of CO oxidation on the reduced CeO2(111)-(3×3) surfaces with 1/9 and 2/9 ML of surface hydroxyl. | |
{kind=link}
Finally, it needs to be noted that CO oxidation on the pristine CeO2(111) surfaces with different surface cells was also calculated and the results are listed in Table S5. No obvious trend of the energy changes can be recognized, suggesting that the size of the surface cell, i.e., CO coverage, itself may have neglectable effect on the calculated energetics.
In summary, CO oxidations on the CeO2(111) surfaces with different reduction degrees implemented by forming surface hydroxyls were theoretically investigated by DFT+U calculations in this work. Energetic components involved in this process, including the adsorption, activation, reaction and bending energies, were determined on the various pristine and reduced surfaces, which largely exhibit linear relationships with the surface reduction degree. In particular, the calculated barrier for the direct reaction between CO and surface lattice O drastically decreases with the increase of surface reduction degree. From electronic analysis, we found that the surface reduction can lead to the occurrence of localized electrons at the surface Ce and affect the charge distribution at surface O. As the result, they become more negatively charged and therefore more active in reacting with CO. We can tentatively suggest that the localized 4f electron reservoir of Ce can act as the "pseudo-anion" at reduced CeO2 surfaces to activate surface lattice O for catalytic oxidative reactions.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for financial support from the National Key R&D Program of China (No. 2018YFA0208602) and National Natural Science Foundation of China (No. 21825301). The authors also thank the National Super Computing Center in Jinan for computing time.
Appendix A. Supplementary dataSupplementarymaterial related tothis article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.08.033.
| [1] |
C.T. Campbell, Science 309 (2005) 713-714. DOI:10.1126/science.1113955 |
| [2] |
(a) X.Q. Gong, L.L. Yin, J. Zhang, et al., Adv. Chem. Eng. 44 (2014) 1-60; (b) D. Ding, X. Li, S.Y. Lai, K. Gerdes, M. Liu, Energy Env. Sci. 7 (2014) 552-575; (c) D.R. Mullins, Surf. Sci. Rep. 70 (2015) 42-85; (d) T. Montini, M. Melchionna, M. Monai, P. Fornasiero, Chem. Rev. 116 (2016) 5987-6041; (e) A. Wang, J. Li, T. Zhang, Nat. Rev. Chem. 2 (2018) 65-81. |
| [3] |
(a) Y. Namai, K. Fukui, Y.J. Iwasawa, Phys. Chem. B 107 (2003) 11666-11673; (b) F. Esch, S. Fabris, L. Zhou, et al., Science 309 (2005) 752-755; (c) M. Nolan, J. Fearon, G. Watson, Solid State Ion. 177 (2006) 3069-3074; (d) H.Y. Li, H.F. Wang, X.Q. Gong, et al., Phys. Rev. B 79 (2009) 193401; (e) B. Chen, Y. Ma, L. Ding, et al., J. Phys. Chem. C 117 (2013) 5800-5810; (f) X.P. Wu, X.Q. Gong, J. Am. Chem. Soc. 137 (2015) 13228-13231; (g) X.P. Wu, X.Q. Gong, Phys. Rev. Lett. 116 (2016) 086102; (h) S. Li, Y. Xu, Y. Chen, et al., Angew. Chem. Int. Ed. 56 (2017) 10761-10765. |
| [4] |
C. Doornkamp, V. Ponec, J. Mol. Catal. Chem. 162 (2000) 19-32. DOI:10.1016/S1381-1169(00)00319-8 |
| [5] |
(a) E. Aneggi, J. Llorca, M. Boaro, A.J. Trovarelli, J. Catal. 234 (2005) 88-95; (b) C. Wang, X.K. Gu, H. Yan, et al., ACS Catal. 7 (2017) 887-891. |
| [6] |
F. Chen, D. Liu, J. Zhang, et al., Phys. Chem. Chem. Phys. 14 (2012) 16573. DOI:10.1039/c2cp41281k |
| [7] |
(a) A. Trovarelli, C. Deleitenburg, G. Dolcetti, J.L. Lorca, J. Catal. 151 (1995) 111-124; (b) H.Y. Kim, H.M. Lee, G. Henkelman, J. Am. Chem. Soc. 134 (2012) 1560-1570; (c) R. Kopelent, J. van Bokhoven, A.J. Szlachetko, et al., Angew. Chem. Int. Ed. 54 (2015) 8728-8731; (d) J.X. Liu, Y. Su, I.A.W. Filot, E.J.M.A. Hensen, J. Am. Chem. Soc. 140 (2018) 4580-4587. |
| [8] |
(a) E. Mamontov, T. Egami, R. Brezny, M. Koranne, S.J. Tyagi, Phys. Chem. B 104 (2000) 11110-11116; (b) M. Zhao, M. Shen, J. Wang, J. Catal. 248 (2007) 258-267; (c) Z. Wu, D.R. Mullins, L.F. Allard, Q. Zhang, L. Wang, Chin. Chem. Lett. 29 (2018) 795-799; (d) Y. Yan, H. Li, Z. Lu, et al., Chin. Chem. Lett. 30 (2019) 1153-1156; (e) X. Guo, Z. Qiu, J. Mao, R. Zhou, J. Power Sources 451 (2020) 227757. |
| [9] |
L. Nie, D. Mei, H. Xiong, et al., Science 358 (2017) 1419-1423. DOI:10.1126/science.aao2109 |
| [10] |
(a) S.D. Senanayake, J. Zhou, A.P. Baddorf, D.R. Mullins, Surf. Sci. 601 (2007) 3215-3223; (b) D. Schweke, L. Shelly, R.B. David, et al., J. Phys. Chem. C 124 (2020) 6180-6187. |
| [11] |
Z. Liu, E. Huang, I. Orozco, et al., Science 368 (2020) 513-517. DOI:10.1126/science.aba5005 |
| [12] |
(a) K.Z. Qi, G.C. Wang, W.J. Zheng, Surf. Sci. 614 (2013) 53-63; (b) K. Qi, F. Zasada, W. Piskorz, et al., J. Phys. Chem. C 120 (2016) 5442-5456; (c) K. Qi, D. Li, J. Fu, et al., J. Phys. Chem. C 118 (2014) 23320-23327. |
| [13] |
(a) L. Song, A. Avorid, G.C. Groenenboom, J. Phys. Chem. A 117 (2013) 7571-7579; (b) H. Zhou, D. Wang, X.Q. Gong, Phys. Chem. Chem. Phys. 22 (2020) 7738-7746. |
| [14] |
(a) Y. Tang, Y.G. Wang, J. Li, J. Phys. Chem. C 121 (2017) 11281-11289; (b) W. Song, L. Chen, J. Deng, et al., Phys. Chem. C 122 (2018) 25290-25300. |