 2021, Vol. 32
2021, Vol. 32 


Efficient delivery of drugs into tumor cells to increase the intracellular drug concentration is one of the important issues in cancer therapy [1-4]. In recent years, a lot of drug nanocarrier systems have been developed to overcome the limitations of conventional chemotherapy, including the poor control over drug release and the distribution of drug in tumor site [5-9]. The nanoscale drug delivery systems (DDSs) can potentially deliver drugs directly into cancerous tissues, and open up entirely new modalities of cancer therapy, such as photodynamic, hyperthermia, and stimulus-responsive treatments [10-14].
Paclitaxel (PTX), a natural anti-tumor drug extracted from Pacific yew, can promote tubulin polymerization of tumor cells, inhibit mitosis and prevent the growth of tumor cells and then play an antitumor effect [15-19]. In the clinical, PTX is mainly used for the treatment of ovarian cancer, breast cancer, and non-small cell lung cancer. Due to the low aqueous solubility of PTX, it is clinically available as Taxol® in a prescription composed of 1:1 of Cremophor EL® (CrEL) (polyoxyethylated castor oil and ethanol). However, side effects can be caused by CrEL including hypersensitivity reactions, nephrotoxicity, neurotoxicity, and cardiotoxicity [18, 20-22].
Drug-conjugated gold nanoparticles (GNPs) have become a new research in the field of DDSs [23, 24]. The GNPs possess their own advantages such as controllable shape and size, large surface area for versatile modification, and biocompatibility and non-toxic nature for biological applications [25-29]. The utilization of GNPs has solved several problems that many drugs encountered in clinical application, such as poor solubility, short half-life, poor bioavailability, and serious side effects. In our previous research, we have designed a type of PTX-conjugated GNPs with PTX located on the surface [30], exhibiting increased solubility and antitumor activity. However, we also found some limitations to apply this conjugate. Firstly, the hydrophobic PTX was located on the surface of gold conjugate. Although the conjugate can be well-dissolved in water, they self-assembled into small clusters composed of ~10 conjugates observed by transmission electron microscopy (TEM). The hydrophobic surface not only increases the system size and tends to absorb more proteins or enzymes in circulation, causing the instability of the conjugates. Secondly, polyethylene glycol (PEG) was connected to 7-hydroxyl group of PTX via a stable ester bond. Although PTX can be released in the presence of esterase in tumor cell cytoplasm, the selectivity of the system is still limited. Based on a series of works we carried out previously, we propose the concept of "structure-efficacy effect" on nanomedicine, especially the drug-conjugated GNPs [31]. It is pointed out that the structure of drug-conjugated GNPs played important roles in modulating their in vitro properties and in vivo performances.
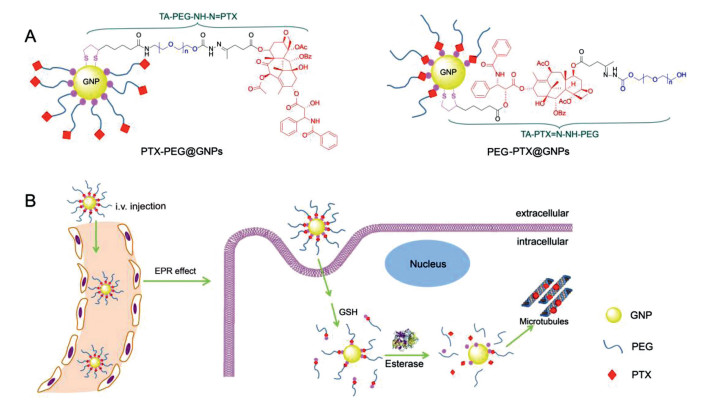
In this work, two new therapeutic ligands, TA-PEG-NH-N=PTX and TA-PTX=N-NH-PEG (Fig. 1, molecular weight 1000 Da), were designed and synthesized to investigate the effect of drug position on property modulation of PTX-conjugated GNPs in vitro and in vivo. In the chemical structures of these ligands, PTX was anchored on one terminal of ligand far from thioctic acid (TA) and inserted between TA and PEG, respectively. After opening the disulfide bond in TA by sodium borohydride, these ligands were easily modified on the surface of GNPs via the S-Au bond. The surface modification of therapeutic ligands provides the conjugates either a hydrophobic PTX shell or a biocompatible PEG outer layer, which offers a platform for investigating the drug position effect. In addition, we also introduced an acid-sensitive hydrazone bond between PTX and PEG in therapeutic ligands. The introduction of hydrazone bond bestows the pH-triggered PTX or PTX-PEG segment release in lysosomes. Combining with the esterase hydrolysis mechanism of PTX-PEG, ligand TA-PTX=N-NH-PEG with two-step drug release process increases the selectivity of drug release under the tumor environments of low pH and high concentration of esterase [32-34].

|
Download:
|
| Fig. 1. Schematic illustration of (A) the chemical structure of therapeutic ligands and their gold conjugates including PTX-PEG@GNPs and PEG-PTX@GNPs, and (B) the cellular uptake of PEG-PTX@GNPs and their two-step drug release process inside tumor cells. | |
{kind=link}
Based on the above platform, in vitro properties and in vivo performance of the as-prepared conjugates have been investigated systematically. Comparing these two gold conjugates, PEG-PTX@GNPs with PTX inserted between GNP and PEG showed an improved solubility, dispersity, tumor inhibition rate, and biosafety. We believe the optimized drug-conjugated GNPs via the "structure-efficacy studies" combining with the selectively tumor microenvironment-stimulated drug release solves the clinical problems of existent pharmaceutics, including their short half-life, poor stability, low bioavailability, toxicity, and so on.
Owing to these strategies, GNPs [35-38] can sufficiently play their enhanced penetration and retention effect, and long circulating drug delivery advantage. The whole drug system can effectively avoid the capture by in vivo P-glycoprotein efflux pump and improve the PTX concentration at the tumor site.
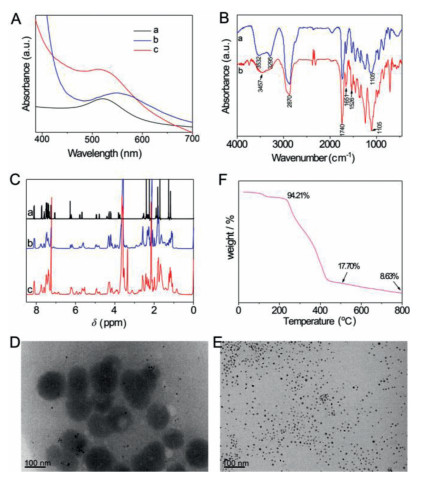
The chemical reaction schemes for synthesizing the therapeutic ligands as well as their 1H and 13C NMR are illustrated in Supporting information. After reduced by sodium borohydride, disulfide bond in TA was opened and formed S-Au bond with GNPs to obtain drug loaded gold conjugates. The resultant two types of PTX-conjugated GNPs were characterized via UV–vis, FT-IR and NMR methods. UV–vis absorption spectrum of GNPs at 518 nm shows that the diameter of the GNPs is about 9 nm (Fig. 2A). An obvious red-shift of GNP peak for PTX-PEG@GNPs could be attributed to the poor solubility and aggregation of PTX-PEG@GNPs, while the slight redshift of peak for PEG-PTX@GNPs indicated their remaining good dispersity in the aqueous solution. According to FT-IR spectra (Fig. 2B), the absorption signals at 3532 cm−1 and 3457 cm−1 indicate the existence of νN-H in gold conjugates. The signals around 3300 cm−1 come from νO-H. The peaks around 1526 cm−1 indicate the existence of νC=C from benzene groups. The peaks situated at 1740 cm−1 and 1651 cm−1 are representative of νC=O from the ester groups. These FT-IR signals show the characteristic infrared signals of both PTX and PEG, which indicate the successful modification of PTX ligands on the surface of GNPs. In addition, 1H NMR spectra were further used to confirm the successful preparation of GNP-paclitaxel conjugates (Fig. 2C). Compared with the PTX, the PEG-PTX@GNPs and PTX-PEG@GNPs exhibit new, broad resonance at 3.6 ppm that corresponds to the protons of PEG, and the characteristic peaks of PTX can also be observed, which mean the existence of both PEG and PTX in the conjugates.

|
Download:
|
| Fig. 2. Characterization of PTX-conjugated GNPs. (A) UV–vis spectra of (a) GNPs, (b) PTX-PEG@GNPs and (c) PEG-PTX@GNPs. (B) FT-IR spectra of (a) PTX-PEG@GNPs and (b) PEG-PTX@GNPs in KBr. (C) 1H NMR spectra of (a) PTX, (b) PTX-PEG@GNPs and (c) PEG-PTX@GNPs. TEM images of (D) PTX-PEG@GNPs and (E) PEG-PTX@GNPs. Scale bar: 100 nm. (F) TGA measurement of the loss of organic material corresponding to the PEG-PTX@GNPs. | |
{kind=link}
The morphologies of PTX-conjugated GNPs were investigated by TEM. As shown in Figs. 2D and E, the dark gold cores are spherical with a diameter of ~9 nm. However, the PTX-PEG@GNP particles are aggregated, consistent with its result in UV–vis spectrum (Fig. 2A, curve b). It is due to the hydrophobic property of PTX located at the outer layer of PTX-PEG@GNPs. On the contrary, the hydrophilic PEG on the surface of the PEG-PTX@GNPs leads to the good dispersion of PEG-PTX@GNPs in aqueous solution. Accordingly, this aggregation and dispersity states of gold conjugates are also reflected in their dynamic light scattering (DLS) data, a larger hydrodynamic average diameter of PTX-PEG@GNPs (353.2±0.4 nm) than that of PEG-PTX@GNPs (300.3±8.5 nm). Moreover, the zeta potential values are 1.4±0.1 mV for PTX-PEG@GNPs and −3.5±0.4 mV for PEG-PTX@GNPs, respectively, confirming that the surfaces of two gold conjugates were modified with different moieties (PTX or PEG).
The advantages of PEG-PTX@GNPs in terms of its physical and chemical properties can be used for further development of drug delivery system. To determine the PTX loading capacity in the PEG-PTX@GNPs, TGA was carried out to evaluate the organic loss. The TGA curves shown in Fig. 2F demonstrate that the composition of the PEG-PTX@GNPs conjugate was 82.3% organic and 17.7% metallic gold after dialysis at 500 ℃. The drug loading capacity (DLC) of PEG-PTX@GNPs was calculated to be 32% by calculating the individual molar mass of PTX and its corresponding ligand are 854 g/mol and 2198 g/mol, respectively.
In addition, the stability of the PEG-PTX@GNPs was evaluated in different environments that can induce the aggregation of unmodified gold particles by measuring UV–vis absorption intensity at 520 nm (Fig. S1 in Supporting information). In buffers with neutral pH values (0.02 mol/L PBS, pH 7.4), the elevated salt concentrations (0.3 mol/L PBS, pH 7.4) and in the presence of 2% serum (0.02 mol/L PBS, pH 7.4, + 2% serum), the relative absorbance of PEG-PTX@GNPs at 10 h was no less than 91.4% of the gold conjugates at 0 h, indicating that the conjugates are stable, due to the covalent linkage in the components of PEG-PTX@GNPs. However, in buffers with low pH values (0.02 mol/L PBS, pH 5.5), PEG-PTX@GNPs are not stable. It can be due to the cleavage of pH-sensitive hydrazone bonds in the therapeutic ligand that released PTX-PEG segment in an acidic environment.
Here, three important parameters influence the drug release of PEG-PTX@GNPs. On the basis of the chemical structure of TA-PTX=N-NH-PEG, free PTX is expected to be released under the synergistic conditions of the different (1) pH value environments in tumor tissue or organelles, (2) glutathione (GSH) concentration and (3) esterase levels inside or outside tumor cells. Under these conditions, in vitro drug release studies of PEG-PTX@GNPs were systematically investigated (Fig. S2 in Supporting information).
Firstly, as DDSs, pH sensitive system has been most widely used due to the lower pH in tumor environments than that in normal tissues caused by the high rate of glycolysis in cancer cells, both in aerobic and anaerobic conditions. Tumors have been demonstrated to exhibit acidic pH values ranging from 5.7 to 7.8, while the pH of normal tissue is 7.4 [39, 40]. Even greater pH differences can be found at the subcellular level; late endosomes and lysosomes have much lower pH, in the range 4.5−5.5. Therefore, pH-sensitive delivery systems are valuable for controlling drug delivery in cancerous diseases. Fig. S2A shows the drug release profiles of PTX in PBS with different pH values. The release of free PTX was increased with the decrease of pH value, due to the cleavage of the pH-sensitive hydrazone bond.
Secondly, the TA-PTX=N-NH-PEG ligands were modified on GNPs based on Au-S groups. The GSH at extracellular and intracellular can function as a reducing agent to exchange the TA-PTX=N-NH-PEG from the surface of GNPs. The GSH-induced drug release from the conjugates was related to the GSH concentration (Fig. S2B). The drug was liberated from the conjugates in the form of ligands, especially in the release media with intracellular GSH levels (10 mmol/L). The cumulative drug release peaked after 70 h at ca. 12%, indicating that the release velocity of the ligands was controlled by the GSH concentration-dependent ligand exchange rate.
Thirdly, pig liver esterase (PLE) was also used to study the drug release of the conjugates, because mammalian esterases are widely distributed throughout the plasma, many organs, and tissues (Fig. S2C). In the presence of PLE, free PTX was slowly dissociated from the conjugates, and the cumulative percentage of PTX release in the presence of PLE was about 12% in 70 h.
Finally, we investigated the drug release in the media meeting the above three conditions at the same time. The drug release from conjugate is still quite low: the cumulative percentage of PTX release was about 14.5% in 70 h (Fig. S2D). It indicates that most of the conjugates remain stable in circulation for long periods of time in vivo. Either low pH, high GSH concentration, or high PLE level, can increase the drug release. However, these pathological environments are not completely overlayered with each other. It is difficult to simulate the drug release process in the body. Therefore, we use the bioactivity of conjugates to substitute the detection of drug content for evaluating their drug release.
Fig. 3A shows the viability of HepG2 cells after treatment with the distinct PTX formulations. Compared to free PTX, the cytotoxicity of PEG-PTX@GNPs was lower in low concentration level. With the increase of PTX concentration, they exhibited nearly the same toxicity. The potential reason is that free PTX is directly dispersed in the solvent and exerted efficacy in the cell. In comparison, the drug release from PEG-PTX@GNPs needs to crack covalent hydrazone bond, S-Au bond and ester bond, which is more selective and safe to normal tissues. To HepG2 tumor cells, PEG-PTX@GNPs also show a high cytotoxicity (IC50 = 1.0 μg/mL), which might be due to the increased cellular uptake (Fig. 3B) and intracellular PTX release via a synergistic manner.

|
Download:
|
| Fig. 3. Cell experiments. (A) In vitro MTT assays to measure the cytotoxicity of (a) PTX and (b) PEG-PTX@GNPs with varying PTX concentrations for 48 h against HepG2 cells. (B) Intracellular PTX content detection of PTX and PEG-PTX@GNPs in HepG2 cells at 4, 8, 12, and 24 h. (C) TEM images of HepG2 cells under the following conditions: cells cultured with GNPs for (a) 4 h and (b) 12 h, or with PEG-PTX@GNPs for (c) 4 h and (d) 12 h. | |
{kind=link}
Detected by high performance liquid chromatography (HPLC) and subsequent correction using intracellular protein content, intracellular drug content was shown in Fig. 3B. PEG-PTX@GNP-treated group showed about two-times higher PTX concentration than the free PTX group in HepG2 cells from 4 h to 24 h and the drug was in the form of PTX. This phenomenon implied the sustained and effective drug release from PEG-PTX@GNPs in tumor cells.
To investigate the cellular distribution inside tumor cells, HepG2 cells were incubated with PEG-PTX@GNPs at a dose of 2.5 μg/mL PTX for 4 h and 12 h. As shown inFig. 3C, both GNPs and PEG-PTX@GNPs are present in the cytoplasm of HepG2 cells after 4 h of incubation. Furthermore, Fig. 3C-b and Fig. 3C-d showed even more nanoparticles localized in the cytoplasm after 12 h of incubation. At this time, cell edges are in irregular state and show projection and depression, indicating that free PTX is released from the nanoparticle and causes damage to the cell.
After intravenous administration of commercial PTX formulation Taxol® and PEG-PTX@GNPs in rats, we characterized the plasma concentration-time profiles of PTX (Fig. S3A in Supporting information). The pharmacokinetic parameters of PTX in the two formulations are presented in Fig. S3B (Supporting information). As shown in Fig. S3A, PEG-PTX@GNPs produced a higher plasma concentration of PTX than the Taxol® group from 5 min to 48 h. For PEG-PTX@GNPs, the area under the plasma concentration-time curve (AUC0−48 h) was 18.06±1.34 μg mL−1kg−1, which is approximately 4.5-fold higher than free PTX (4.10±0.55 μg mL−1kg−1), and the corresponding total body clearance (CL) was 0.21±0.03 L/h, which was 8-fold lower than Taxol® (1.72±0.22 L/h). These data suggest that the conjugate group displayed slower elimination of PTX, longer plasma half-life (t1/2) and longer mean residence time (MRT) compared to Taxol®, confirming the longer circulation characteristics of PEG-PTX@GNPs in blood.
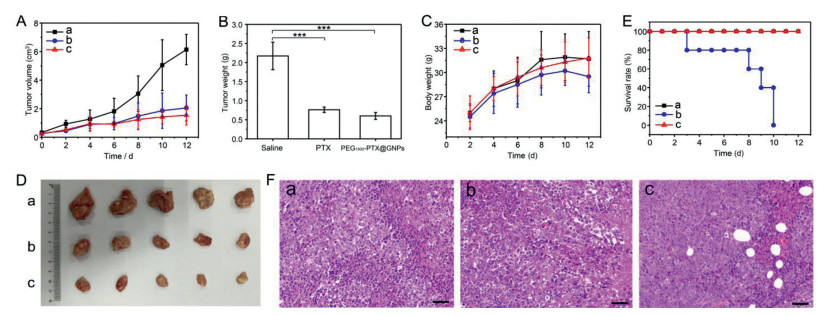
Moreover, antitumor efficacy was evaluated in ICR mice bearing Heps tumors. Changes in tumor volume, final tumor weight, tumor images and the body weight of mice treated with saline, Taxol®, and PEG-PTX@GNPs are presented in Figs. 4A-D. Tumor growth was significantly suppressed in the group treated with PEG-PTX@GNPs compared to saline, even better than Taxol®. Although the body weight of PEG-PTX@GNPs group (31.8±2.5 g) was as heavy as saline group, exceeded that of the Taxol® groups, it had the smallest tumor size and tumor weight. These results indicate improved quality of life in mice treated with the conjugate.

|
Download:
|
| Fig. 4. In vivo antitumor efficacy of (a) saline, (b) Taxol® and (c) PEG-PTX@GNPs in Heps tumor xenograft ICR mouse models. (A) tumor volume changes, (B) tumor tissue weight 10 days post-injection (***P < 0.001), (C) body weight changes, (D) images of the tumor tissues excised from mice on day 10, (E) survival rates of the three groups of tumor-bearing mice, (F) H & E stained tumor tissue sections from control and test groups at 10 days post-injection (Scale bar: 50 μm). | |
{kind=link}
As for survival rates of the three groups (Fig. 4E), no mice died in the PEG-PTX@GNPs or saline-treated group (n=5) after 12 days, whereas no one was survival in Taxol®-treated group. Great systemic toxicity of Taxol® in vivo can be seen from this result, but treatment with PEG-PTX@GNPs can effectively reduce this side effect and prolong their life. This is due to the increased tumor selectivity produced by PEG-PTX@GNPs. The enhanced therapeutic efficacy of PEG-PTX@GNPs is also owing to long circulation and the successful passive tumor targeting of the conjugates in tumor tissues.
The PTX concentration in different tissues of tumor bearing mice treated with Taxol® decreased gradually with time going. However, PEG-PTX@GNPs kept relatively high PTX level. This result provides additional evidence for the high stability and sustained drug release of the conjugates observed in in vitro drug release assays. For the maximum PTX amount in heart, liver, spleen and lung, the Taxol® group are much higher than that of PEG-PTX@GNPs (Fig. S4 in Supporting information). In addition, the PTX concentration in the tumor tissues from the PEG-PTX@GNPs group was higher, and it was eliminated more slowly than was observed for the Taxol® group.
To determine the extent of tumor damage in response to concentrated distribution of the conjugate and the high level of Taxol® released from it, we performed histological examination of H&E stained tumor tissue sections at 12-day post-injection (Fig. 4F). Almost no damage was observed in tumors from the saline-treated group, but significant necrosis was present in both the Taxol® and PEG-PTX@GNPs-treated groups. These data further confirmed the therapeutic effect of the nano-prodrug. Therefore, the pH-sensitive and controlled drug release in tumor tissues ensured that higher amounts of PTX at tumor site, resulting in increased therapeutic efficacy and anti-tumor efficiency.
In summary, we have developed two types of PTX-conjugated GNPs for investigating their in vitro properties and in vivo performance affected by the drug position in the therapeutic ligands. In these systems, GNPs were selected as the nano-drug delivery carrier and two therapeutic ligands (TA-PEG-NH-N=PTX and TA-PTX=N-NH-PEG) consisted of PTX and PEG were synthesized for conjugating to the surface of GNPs. After investigating their solubility, drug loading, surface charge, and particle size, we found the TA-PTX=N-NH-PEG modified GNPs (PEG-PTX@GNPs) showed optimized properties for drug delivery. The PEG-PTX@GNPs exhibit high aqueous solubility, good biocompatibility, good stability, good anticancer activity, controlled drug release, and enhanced therapeutic efficacy. The advantages of the optimized drug loading system can be potentially used as a promising approach for in vivo tumor therapy.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was supported by grants from the National Natural Science Foundation of China (Nos. 31870946, 31470916, 31500769 and 21705165), the Funding of Double First-rate Discipline Construction of China (No. CPU2018GF07), Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Fundamental Research Funds for the Central Universities (Nos. 2015PT036, 2016PT014, and 3011900159).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.08.044.
| [1] |
A. El-Aneed, J. Control. Release 94 (2004) 1-14. DOI:10.1016/j.jconrel.2003.09.013 |
| [2] |
G.A. Hughes, Nanomedicine 1 (2005) 22-30. DOI:10.1016/j.nano.2004.11.009 |
| [3] |
K. Ou, X. Xu, S. Guan, et al., Adv. Funct. Mater. 30 (2020) 1907857. DOI:10.1002/adfm.201907857 |
| [4] |
B. Wang, P. Lv, H. Cai, et al., J. Biomed. Nanotechnol. 15 (2019) 1897-1908. DOI:10.1166/jbn.2019.2833 |
| [5] |
J.L. Arias, Curr. Med. Chem. 19 (2012) 3069-3069. DOI:10.2174/092986712800784649 |
| [6] |
J.O. Kim, C.S. Yong, J.Y. Choi, R.K. Thapa, J. Pharm. Invest. 46 (2016) 325-339. DOI:10.1007/s40005-016-0252-1 |
| [7] |
K.R. Vega-Villa, J.K. Takemoto, J.A. Yanez, et al., Adv. Drug Deliver. Rev. 60 (2008) 929-938. DOI:10.1016/j.addr.2007.11.007 |
| [8] |
P.E. Saw, X. Xu, M. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 6249-6252. DOI:10.1002/anie.201916574 |
| [9] |
Y. Zheng, X. You, S. Guan, et al., Adv. Funct. Mater. 29 (2019) 1808646. DOI:10.1002/adfm.201808646 |
| [10] |
D. Farrell, K. Ptak, N.J. Panaro, P. Grodzinski, Pharmaceut. Res. 28 (2011) 273-278. DOI:10.1007/s11095-010-0214-7 |
| [11] |
S. Fitzpatrick, Nanomedicine 8 (2013) 326-326. |
| [12] |
X.M. Liu, B. Yang, Y.L. Wang, J.Y. Wang, Chem. Mater. 17 (2005) 2792-2795. DOI:10.1021/cm0479335 |
| [13] |
L. Qiu, C. Zheng, Y. Jin, K. Zhu, Expert Opin. Ther. Pat. 17 (2007) 819-830. DOI:10.1517/13543776.17.7.819 |
| [14] |
S. Singh, J. Nanosci. Nanotechnol. 10 (2010) 7906-7918. DOI:10.1166/jnn.2010.3617 |
| [15] |
A. Gollerkeri, L. Harrold, M. Rose, D. Jain, B.A. Burtness, Int. J. Cancer 93 (2001) 139-141. DOI:10.1002/ijc.1295 |
| [16] |
J. Li, F. Wang, D. Sun, R. Wang, J. Drug Target. 24 (2016) 590-602. DOI:10.3109/1061186X.2016.1154561 |
| [17] |
D.A. Yardley, J. Control. Release 170 (2013) 365-372. DOI:10.1016/j.jconrel.2013.05.041 |
| [18] |
H. Li, J. Li, X. He, et al., Chin. Chem. Lett. 30 (2019) 1083-1088. DOI:10.1016/j.cclet.2019.01.003 |
| [19] |
M. Sang, Z. Zhang, F. Liu, et al., J. Biomed. Nanotechnol. 14 (2018) 477-495. DOI:10.1166/jbn.2018.2541 |
| [20] |
N.M. Chu, C. Hsieh, W. Fang, S. Chang, Z. Chu, J. Clin. Oncol. 22 (2004) 7260-7260. DOI:10.1200/jco.2004.22.90140.7260 |
| [21] |
E.K. Rowinsky, E.A. Eisenhauer, V. Chaudhry, S.G. Arbuck, R.C. Donehower, Semin. Oncol. 20 (1993) 1-15. |
| [22] |
N. Yu, J. Li, P.K. Singh, et al., J. Biomed. Nanotechnol. 15 (2019) 2251-2261. DOI:10.1166/jbn.2019.2847 |
| [23] |
W. Yu, M. Shevtsov, X. Chen, H. Gao, Chin. Chem. Lett. 31 (2020) 1366-1374. DOI:10.1016/j.cclet.2020.02.036 |
| [24] |
M. Malathi, W. Christian, X. Chenjie, Nano-Micro Lett. 10 (2018) 10. DOI:10.1007/s40820-017-0166-0 |
| [25] |
T. Cui, J.J. Liang, H. Chen, et al., ACS Appl. Mater. Interfaces 9 (2017) 8569-8580. DOI:10.1021/acsami.6b16669 |
| [26] |
J.R. Nicol, D. Dixon, J.A. Coulter, Nanomedicine 10 (2015) 1315-1326. DOI:10.2217/nnm.14.219 |
| [27] |
G.F. Paciotti, L.D. Myer, T.H. Kim, et al., Clin. Cancer Res. 7 (2001) 3673S-3674S. |
| [28] |
F. Qiu, Y. Wang, D. Yang, T. Zhang, J. Pan, J. Control. Release 259 (2017) e34. |
| [29] |
S. Yang, L. Zhou, Y. Su, R. Zhang, C.-M. Dong, Chin. Chem. Lett. 30 (2019) 187-191. DOI:10.1016/j.cclet.2018.02.015 |
| [30] |
Y. Ding, Y.Y. Zhou, H. Chen, et al., Biomaterials 34 (2013) 10217-10227. DOI:10.1016/j.biomaterials.2013.09.008 |
| [31] |
W. Wang, J. Wang, Y. Ding, J. Mater. Chem. B 8 (2020) 4813-4830. DOI:10.1039/C9TB02924A |
| [32] |
Y. Jin, Y. Huang, H. Yang, G. Liu, R. Zhao, Chem. Commun. 51 (2015) 14454-14457. DOI:10.1039/C5CC05184C |
| [33] |
P. Qi, Y. Bu, J. Xu, et al., Colloid Polym. Sci. 295 (2017) 1-12. DOI:10.1007/s00396-016-3968-6 |
| [34] |
O. Sedlacek, B.D. Monnery, J. Mattova, et al., J. Control. Release 259 (2017) e100-e101. |
| [35] |
K. Haume, S. Rosa, S. Grellet, et al., Cancer Nanotechnol. 7 (2016) 8. DOI:10.1186/s12645-016-0021-x |
| [36] |
Z. Liu, F. Pu, J. Liu, et al., Nanoscale 5 (2013) 4252-4261. DOI:10.1039/c3nr00491k |
| [37] |
C.R. Patra, R. Bhattacharya, D. Mukhopadhyay, P. Mukherjee, Adv. Drug Deliver. Rev. 62 (2010) 346-361. DOI:10.1016/j.addr.2009.11.007 |
| [38] |
E. Shamaeli, N. Alizadeh, Colloid Surf. B 126 (2015) 502-509. DOI:10.1016/j.colsurfb.2015.01.003 |
| [39] |
K. Engin, D.B. Leeper, J.R. Cater, et al., Int. J. Hyperthermia 11 (1995) 211-216. DOI:10.3109/02656739509022457 |
| [40] |
M. Stubbs, P.M.J. McSheehy, J.R. Griffiths, C.L. Bashford, Mol. Med. Today 6 (2000) 15-19. DOI:10.1016/S1357-4310(99)01615-9 |