 2021, Vol. 32
2021, Vol. 32 


 Junsong Zhong obtained his BSc degree in chemical biology from Xiamen University in 2019. Then he continued his research at Fuzhou University under the guidance of Prof. Ke-Yin Ye. His research interests focuse on electrochemical synthesis;
Junsong Zhong obtained his BSc degree in chemical biology from Xiamen University in 2019. Then he continued his research at Fuzhou University under the guidance of Prof. Ke-Yin Ye. His research interests focuse on electrochemical synthesis; |
 Yi Yu obtained her master degree in organic chemistry in 2019 from Jiangxi Normal University. Then she moved to the Fuzhou University for her doctoral studies under the guidance of Prof. Ke-Yin Ye. Her research interests focuse on electrochemical synthesis;
Yi Yu obtained her master degree in organic chemistry in 2019 from Jiangxi Normal University. Then she moved to the Fuzhou University for her doctoral studies under the guidance of Prof. Ke-Yin Ye. Her research interests focuse on electrochemical synthesis; |
 Dongliang Zhang is an undergraduate student at Fuzhou University. At present, he is studying and working in Prof. Ke-Yin Ye's research group. His research focuses on electrochemical synthesis; Dongliang Zhang is an undergraduate student at Fuzhou University. At present, he is studying and working in Prof. Ke-Yin Ye's research group. His research focuses on electrochemical synthesis; |
 Prof. Dr. Keyin Ye obtained his bachelor degree from Xiamen University (mentors: Prof. Pei-Qiang Huang and Prof. Hong-Kui Zhang) and PhD from Shanghai Institute of Organic Chemistry (mentors: Prof. Li-Xin Dai and Prof. Shu-Li You). He then conducted his postdoctoral research with Prof. Gerhard Erker (Universität Münster) and Prof. Song Lin (Cornell University), respectively. Since June 2019 he started his independent career at Fuzhou University as a Minjiang Professor. His research interests include organometallics, asymmetric catalysis and electrosynthesis. Prof. Dr. Keyin Ye obtained his bachelor degree from Xiamen University (mentors: Prof. Pei-Qiang Huang and Prof. Hong-Kui Zhang) and PhD from Shanghai Institute of Organic Chemistry (mentors: Prof. Li-Xin Dai and Prof. Shu-Li You). He then conducted his postdoctoral research with Prof. Gerhard Erker (Universität Münster) and Prof. Song Lin (Cornell University), respectively. Since June 2019 he started his independent career at Fuzhou University as a Minjiang Professor. His research interests include organometallics, asymmetric catalysis and electrosynthesis. |
Cobalt has received considerable attention of chemists in organic synthesis owing to its earth-abundant, low-cost, and non-toxic characters [1]. One of the most prominent reactions catalyzed by cobalt probably is the industry-scale Fisher-Tropsch process which converts carbon monoxide and dihydrogen (syngas) into hydrocarbons of various molecular weights [2, 3]. In addition, vitamin B12, the only naturally occurring, cobalt-containing organometallic complex, is also found to be capable to mediate diverse organic transformations both in vivo and in vitro [4, 5]. Being the most sustainable group 9 metal (relative geosphere abundance: Co: Rh: Ir=104: 5:1), there is an increasing demand of using cobalt to replace its noble congeners not only in the known conventional organic reactions but also in the searching of unprecedented and interesting reactivity.
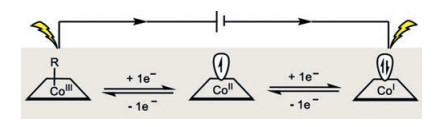
Cobalt accommodates a variety of oxidation states such as -1, 0, +1, +2 and +3. The low-valent cobalt species (-1 and 0) are frequently found in some well-known organometallic complexes and starting materials [6, 7]. On the other hand, the other oxidation states (+1, +2 and +3) are preferentially engaged in stoichiometric as well as catalytic reactions [8, 9]. Similar to the behaviors of rhodium and iridium, the formal +1/+3 oxidation states of cobalt (usually generated from one-electron reduction or one-electron oxidation of its +2 oxidation state form) account for a majority of the diverse redox reactivity observed. Therefore, a very broad range of reactivity including nucleophilic, electrophilic, as well as radical pathways could be readily accessible (Scheme 1). For instance, the competent oxidizing capacity of cobalt(Ⅲ) makes it perfect catalyst in C–H functionalization and oxidative catalysis. The low-valent cobalt(Ⅰ) species, on the other hand, readily activates organohalides either via an oxidative addition or a single-electron transfer process for downstream transformations.

|
Download:
|
| Scheme 1. Merger of cobalt catalysis and electrochemistry. | |
Therefore, electrochemistry would be the perfect partner of cobalt in organic synthesis [10-16]. The employment of the traceless electrons in the redox processes obviates the need of external oxidant and reductant. In addition, the selectivity could be further controlled by the fine-tuning of the applied potential bias. Both features of electrochemistry ensure the much more enhanced compatibility of redox functionalities. On the other hand, cobalt species itself could act as the redox mediator so the shortcomings frequently associated with direct electrolysis including the over oxidation/reduction and electrode passivation could be perfectly avoided [17-19].
Indeed, the constructive merger of cobalt catalysis and electrochemistry, i.e., cobaltaelectrocatalysis [20, 21] has emerged as a very powerful tool for organic chemists to expand current synthetic boundary for the searching of new reactivity [22, 23]. This review aims to provide a comprehensive survey of merging cobalt catalysis and electrochemistry in organic synthesis. To this end, a thorough discussion of the reaction scope, mechanism as well as innovative applications of both the oxidative and reductive cobaltaelectrocatalysis is covered.
2. Oxidative cobaltaelectrocatalysis 2.1. Cobaltaelectro-catalyzed C–H functionalizationCobalt-catalyzed C–H functionalization typically requires the use of excess amount of strong oxidizing agents at high temperature. Therefore, functionalities sensitive to strong oxidants and heat are generally not well tolerated. To this end, Ackermann and co-workers pioneeringly merged the cobalt-catalyzed C–H oxygenation with the electrochemistry under very mild reaction conditions [24]. The frequently used oxidant (e.g., silver salts) in the C–H functionalization catalysis was elegantly replaced by the green and sustainable electricity. Among various N-substituted amides evaluated as the directing groups, the bidentate pyridine -N-oxide motif (PyO) was found to be particularly effective in this transformation. The reaction was initially proposed to proceed a Co(Ⅱ/Ⅲ/Ⅰ) mechanism (Scheme 2, left) where it begins with the anodic oxidation of Co(Ⅱ) 4 to Co(Ⅲ) 5 (E=1.19 V vs. SCE) in the presence of one molecule of alcohol. A facile carboxylate-assisted C–H activation then gives the intermediate 6. Following a reductive elimination and ligand dissociation, the desired C–H oxygenation product 3 is formed accompanied by the formation of the Co(Ⅰ) 8, which would be anodically re-oxidized to the catalytic active Co(Ⅲ) species.

|
Download:
|
| Scheme 2. Proposed mechanisms of cobaltaelectro-catalyzed C-H oxygenation [24]. | |
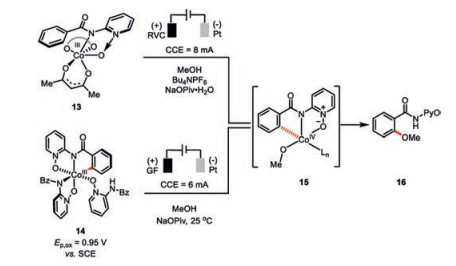
Interestingly, recent mechanistic studies have unraveled another potentially viable Co(Ⅲ/Ⅳ/Ⅱ) reaction pathway (Scheme 2, right), in which cobalt(Ⅱ) 4 was suggested to be anodically oxidized to cobalt(Ⅲ) 9 then cobalt(Ⅳ) 11 [25]. An unusual radical type, carboxylate-assisted HAT (hydrogen-atom transfer) process then proceeds during the formation of complex 12 based on the DFT calculations, which is followed by a ligand exchange and reductive elimination to release the C–H oxygenation product 3 and regenerate the cobalt(Ⅱ) catalyst. Experimental observation of the competence of preformed cobalt(Ⅲ) species 13 in the C–H oxygenation only under electrochemical setting provided strong support of cobalt species in higher oxidation state being the actual catalyst (Scheme 3).

On the other hand, the anodic oxidation of the mixture of cobalt and compound 1 in the absence of alcohol only led to the isolation of the cyclometalated cobalt species 14 comprised of extra two molecules of compound 1 bonding to the cobalt(Ⅲ) center (Scheme 3) [26]. Cyclic voltammetry studies indicated further oxidation of such a Co(Ⅲ) intermediate (=0.95 V vs. SCE) in the presence of alcohol would result in the rare observed Co(Ⅳ) species 15 responsible for the formation of desired C–H oxygenation product 16. In addition, the resulting Co(Ⅱ) species was also identified experimentally. Therefore, another oxidation-induced reductive elimination within a cobalt(Ⅲ/Ⅳ/Ⅱ) mechanism was also proposed for such a cobaltaelectrocatalysis.
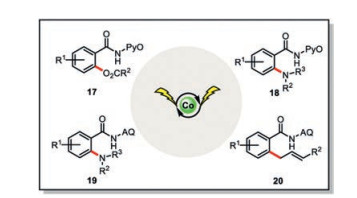
The cobaltaelectrocatalysis quickly found diverse applications in the C–H functionalization [27, 28]. Further extension of this concept to the C–H acyloxylation with carboxylic acid (17) [29], C–H amination with free amines (18, 19) [30, 31], and allylic C–H functionalization of terminal olefins (20) [32] were also realized (Scheme 4). Compared with the conventional chemical approaches, such a cobaltaelectrocatalysis merits from the obviation of external oxidant, room temperature operation, excellent functional compatibility, good scalability, as well as the possibility of reaction being run in a biomass-derived renewable solvent. All these features make this electrochemical approach appealing to the potentially industrial scale applications.

|
Download:
|
| Scheme 4. Cobaltaelectro-catalyzed C-H acyloxylation, amination and allylic C-H functionalization [27-32]. | |
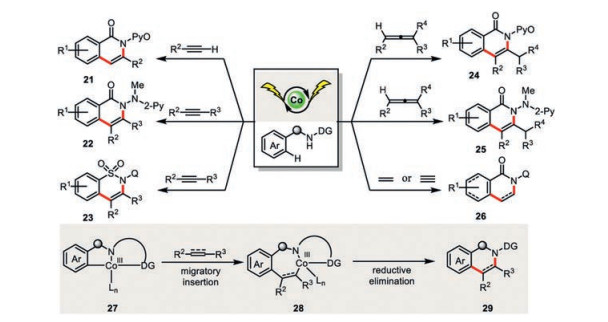
The electrochemically generated cyclometalated cobalt(Ⅲ) species is also a very good catalyst for the other C-C bond(s) forming reactions (Scheme 5). Indeed, the introduction of a variety of π-bond acceptors including substituted terminal alkynes (21) [33, 34], internal alkynes (22, 23) [35, 36] and allenes (24, 25) [37, 38] could all lead to the C–H/N-H functionalization products in good to excellent yields. Lei and co-workers could even extend such a cobaltaelectro-catalyzed C-H/N-H cyclization to the ethylene and acetylene (26) [39]. Mechanistically, the C-Co bond in the cyclometalated cobalt species 27 would undergo a facile migratory insertion across the π-bond to the form the first C-C bond (28). Subsequent C-N reductive elimination furnishes the desired annulated product 29.

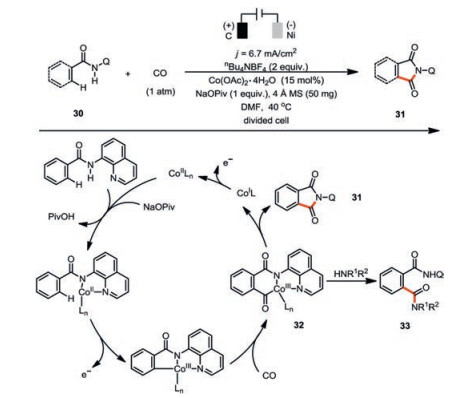
Lei and co-workers elegantly showed that carbon monoxide is another interesting π-bond acceptor in such a cobaltaelectrocatalysis (Scheme 6) [40]. Both the X-ray absorption near edge structure spectroscopy (XANES) and the cyclic voltammetry studies were supportive of Co(Ⅲ) species being generated through the anodic oxidation of the coordinated Co(Ⅱ) species. However, the ligand exchange of N-(quinolin-8-yl)benzamide 30 to Co(Ⅱ) was assigned to facilitate the anodic oxidation, which is in contrast to Ackermann's work where the pivalate ligand played the same role. It seems the substrates with different directing groups would profoundly change the fundamental steps in the reaction mechanism. Analogously to the reaction mechanism of the C-H/N-H cyclization of alkynes, the 1, 1-migratory insertion of C-Co bond to the carbon monoxide forges the first C-C bond (32). Intramolecular C-N bond reductive elimination would then deliver the [4+1] annulated product 31. In the presence of external amine nucleophile, intermolecular reaction dominated and thus formation of the intermolecular carbonylation product 33 was observed. Since the cobalt catalyst is easily reduced at the Ni cathode, a divided cell set-up is necessary. Similarly, the same reactivity could also be observed for substituted isocyanides [41, 42].

|
Download:
|
| Scheme 6. Cobaltaelectro-catalyzed C-H carbonylation [40]. | |
2.2. Cobaltaelectro-catalyzed diverse oxidative transformations
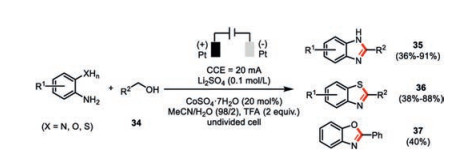
Huang and co-workers developed a cobalt-catalyzed electrochemical synthesis of a diverse array of 2-substituted benzimidazoles 35, benzothiazoles 36, and benzoxazole 37 in good to excellent yields with a wide range of substrate scope (Scheme 7) [43]. The presence of small amount of water was found to be beneficial to the reaction yield. Cyclic voltammetry studies further indicated that the addition of trifluoroacetic acid profoundly inhibited the anodic oxidation of benzylic alcohols (Ep=1.78 V vs. SCE) as well as 1, 2-phenylenediamine (Ep=0.45 V vs. SCE). The authors proposed the cobalt(Ⅱ) sulfate is initially oxidized to Co(Ⅲ) species (Ep=2.04 V vs. SCE), which readily oxidizes benzylic alcohol to the corresponding benzyl aldehyde. Condensation of this aldehyde and 1, 2-phenylenediamine would lead to the cyclic dihydro benzimidazole, additional oxidation of which should render the formation of benzimidazole. Control experiments elucidated the key roles of both the cobalt catalyst as well as the molecule oxygen in the oxidation step(s).

|
Download:
|
| Scheme 7. Cobaltaelectro-catalyzed preparation of benzimidazoles, benzothiazoles, and benzoxazole [43]. | |
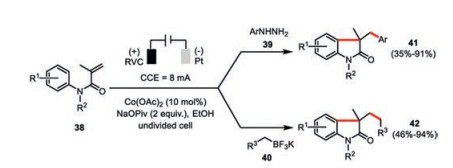
The electrochemically generated high valent cobalt(Ⅲ) species was also demonstrated to be capable to facilitate the efficient preparation of functionalized oxindoles from N-arylacrylamides 38 via cascade radical reactions (Scheme 8) [44]. Both the arylhydrazines 39 and potassium alkyltrifluoroborates 40 were employed as the potential radical precursors. The involvement of radical intermediates was supported by the TEMPO inhibition experiments. Gas-chromatographic headspace analysis also confirmed the formation of molecular nitrogen and hydrogen as the byproducts in the reaction with arylhydrazines as well as molecular hydrogen in the reaction with potassium alkyltrifluoroborates.

|
Download:
|
| Scheme 8. Cobaltaelectro-catalyzed preparation of substituted oxindoles [44]. | |
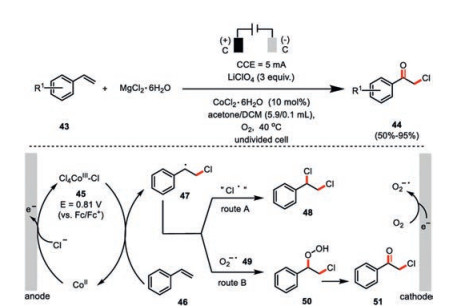
To overcome the time-, labor-and energy-consuming conventional yield-based chemical trial-and-error reaction condition screening, Chen and co-workers applied a potential-based cyclic voltammetry for the optimization of reaction conditions in the cobalt-catalyzed oxychlorination of olefins (Scheme 9) [45]. UV–vis spectroscopy and CV titration experiments indicated the in situ formed catalytically active species to be [Co(Ⅲ)Cl5] 45. Such a high-valent cobalt species is capable to deliver a formal chlorine radical to the styrene 46 leading to a new transient carbon-center radical 47. In the absence of oxygen, such an intermediate is quickly quenched by 45 as being the chlorine source to form dichlorination product 48. In contrast, molecule oxygen could be readily reduced at the cathode to form the persistent superoxide ion 49 (half-life, 30 min. at ambient temperature), which would selectively couple with the previously formed transient carbon-center radical 47 into ultimately formation of the oxychlorination compound 51.

|
Download:
|
| Scheme 9. Cobaltaelectro-catalyzed oxychlorination of olefins [45]. | |
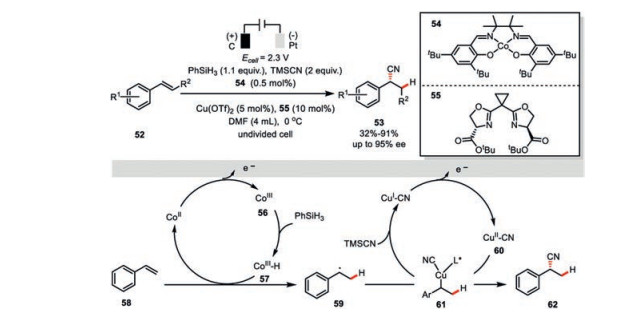
DiStasio, Lin, and their co-workers developed an elegant electrochemical Co/Cu dual-catalyzed highly asymmetric hydrocyanation of olefins (Scheme 10) [46]. This innovative combination of bimetallic catalysis and electrochemistry embraced a very broad array of terminal and internal olefins with diverse functionalities. Delicate balancing the rates of both the HAT and cyanation events was achieved by the fine-tuning of applied potential as well as the catalytic loading of each component, which was requisite for the optimal reactivity as well as selectivity. In the direct comparison, reactions using chemical oxidants only led to neither low yields and/or low enantioselectivities, highlighting the superior capability of electrochemistry in the reactivity and selectivity control.

|
Download:
|
| Scheme 10. Electrochemical Co/Cu dual-catalysis for the hydrocyanation of olefins [46]. | |
With respect to mechanism, the electrochemically generated Co(Ⅲ) species 56 is initially transformed to Co(Ⅲ)-H 57 upon treatment with phenyl silane, which would readily add across the double bond of styrene 58 to deliver a new benzyl radical 59. Interception of this transient intermediate by another anodically generated Cu(Ⅱ)-CN complex 60 then gives rise to the alkyl Cu(Ⅲ) species 61, which should render the formation of the desired hydrocyanation product 62 via a reductive elimination. Their new chiral bisoxazoline ligand 55 featured the uncommon ester groups, which functionalities were proposed to provide attractive noncovalent interactions to stabilize the enantio-determining transition state of the key C-CN bond formation. Such a rational is consistent with both the experiment observation as well as the DFT calculations.
3. Reductive cobaltaelectrocatalysis 3.1. Cobaltaelectro-catalyzed cross-coupling reactionsCobalt-catalyzed cross-coupling reactions are generally between one electrophile and one nucleophile, most representatively, organozinc reagent [47]. Recently, the transition-metal (nickel, palladium etc.) catalyzed bis-electrophile cross-coupling reactions are becoming very economic and promising protocols as which obviate the need of pre-formation of previously requisite nucleophile [48]. However, cobalt is not a good choice to catalyze this type of transformation probably due to its relatively low catalytic reactivity.
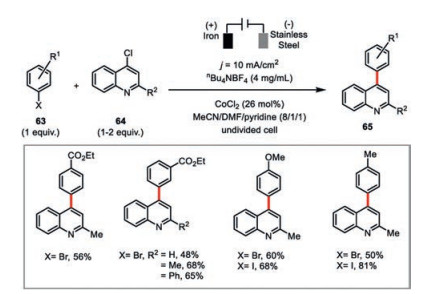
Gosmini and co-workers developed a cobalt(Ⅱ) chloride-catalyzed electrochemical cross-coupling between functionalized phenyl halides 63 and 4-chloroquinoline derivatives 64 in satisfactory to high yields (Scheme 11) [49]. Generally, the yields of cross-coupling products using aryl iodides are higher than aryl bromides. The presence of pyridine to stabilize the electrochemically reduced low-valent cobalt species was essential to the high reaction efficiency. In addition, the fact that only iron but no other metallic anode was capable to facilitate such a transformation indicated the in situ generated ferrous salt during the electrolysis should be involved in this cross-coupling reaction.

|
Download:
|
| Scheme 11. Cobaltaelectro-catalyzed aryl-heteroaryl cross-coupling [49]. | |
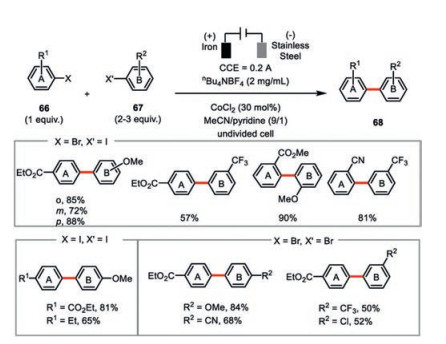
A more general electroreductive cobalt-catalyzed cross-coupling of aryl halides towards the synthesis of unsymmetrical biaryls 68 was advanced by the same group (Scheme 12) [50]. This electrochemical cross-coupling protocol embraced a broad amenable combination of aryl halides in good to excellent yields. Consistent with their previous study, the iron anode and pyridine were both indispensable to the success of this reaction. Interestingly, the attempt to replace pyridine by 2, 2′-bipyridine only led to the formation of dehalogenation by-products (ArH). The use of excess amount of the more reactive coupling elements (e.g., aryl iodides vs. aryl bromide) dramatically enhanced the reaction yields. In most cases, an appreciable amount of dimerization by-products from the more reactive aryl halides formed but luckily not affecting the isolation of the desired cross-coupled, unsymmetrical biaryls.

|
Download:
|
| Scheme 12. Cobaltaelectro-catalyzed aryl-aryl cross-coupling [50]. | |
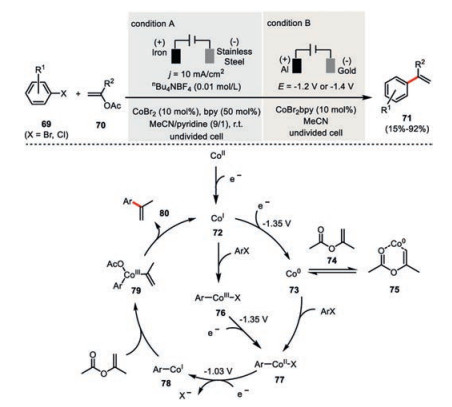
Gosmini's cobaltaelectro-catalyzed cross-coupling protocol could also be well extended to aryl halides 69 and vinyl acetates 70 providing a multitude of functionalized styrenes 71 (Scheme 13, condition A) [51]. Interestingly, in this case the stoichiometric amount of bipyridine was necessary to ensure high reaction yields. The cathodic potential during the constant current electrolysis remained at ~ -1.6 V vs. SCE. This observation is also consistent with the thorough mechanistic investigations by Labbé and Buriez (Scheme 13, condition B) [52]. Examination of the electrochemical behavior of CoBr2(bpy) complex indicated both the low-valent Co(Ⅰ) 72 and Co(0) 73 species should be generated at the same time during the electrolysis and the Co(0) 73 is responsible for the activation of the strong aromatic C-Cl bonds. The complexation of isopropenyl acetate 74 helps to stabilize the Co(0) species 75 but competes with the oxidation event of aryl halide. Experimentally, the reduction potential needed to be more negative than -1.4 V to ensure the efficient reduction of both the Co(Ⅲ) 76 to Co(Ⅱ) 77 and Co(Ⅰ) 72 to Co(0) 73 with both reduction potentials being the same -1.35 V. An additional reduction process then transfers the arylcobalt(Ⅱ) 77 to arylcobalt(Ⅰ) intermediate 78 (E1/2=-1.03 V vs. SCE), which would undergo oxidative addition with the vinylic acetate and then reductive elimination to deliver the cross-coupling product 80.

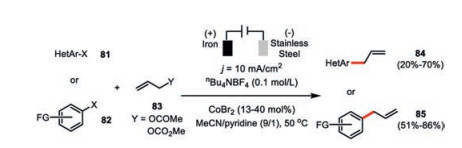
The closely related electrochemical cobalt-catalyzed reductive cross-coupling reaction between aryl halides 81, 82 and allylic acetates 83 was also developed by Gosmini and co-workers (Scheme 14) [53, 54]. While the same transformation under chemical reaction conditions required the judicious choice of appropriate reducing agent (zinc for aryl bromides and manganese for aryl chlorides), the electrochemical variant was free of such a limitation with electron being the traceless and easily tunable reducing agent. The electrolysis was run under constant current control (0.2 A, j =10 mA/cm2) with the similar cathodic potential (-1.1 V vs. SCE) close to the reduction potential of CoX2-pyridine in acetonitrile. The pyridine ligation to cobalt again was indispensable to the reactivity as only a small amount of allylation product could be detected in its absence. While aryl bromides giving the optimal results, aryl chlorides activated by an electron-withdrawing group were also amenable. In contrast, electron-rich aryl chlorides suffered from massive direct reducing by-product (ArH) formation. In addition, aryl iodides were not good choices due to their high reactivity towards the undesired electrochemical aryl dimerization.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Organic zinc compounds are among the most useful reagents in organic synthesis due to their superior reactivity and broad functional compatibility. Typically, alkyl zinc reagents are conveniently prepared from the reaction of alkyl iodides with zinc dust or Rieke zinc while the analogous preparation of aryl zinc compounds requires ultrasonic activation or polar solvent. If starting from the less reactive aryl bromides or chlorides, one needs to firstly prepare their aryl lithium or magnesium compounds then carry out the transmetalation with zinc halides.
To circumvent such a limitation, Gosmini and co-workers developed a cobalt-catalyzed electrochemical procedure of zinc reagents from aryl halides 86 (bromides or chlorides) by using a zinc sacrificial anode (Scheme 15) [55]. The addition of catalytic amount of zinc halides not only increased the reaction yields but also obviated the need of supporting electrolyte. The cathodic potential (constant between -1.1 and -1.3 V vs. SCE) during the electrolysis was well consistent with the one needed for the formation of the Co(Ⅰ) complex from CoX2-pyridine. Therefore, the Co(Ⅰ) generated from the cathodic reduction should be involved in the catalytic formation of zinc compounds. These electrochemically generated zinc reagents 87 were good candidates in the diverse transformations such as direct iodination with iodine (88), palladium-catalyzed cross-coupling reaction (89), cobalt-catalyzed acylation (90) [56] as well as copper-mediated functionalization of pyridine (91) [57].

|
Download:
|
| Scheme 15. Cobaltaelectro-catalyzed in situ generation of organozinc reagents for downstream derivatization [55-57]. | |
{kind=link}
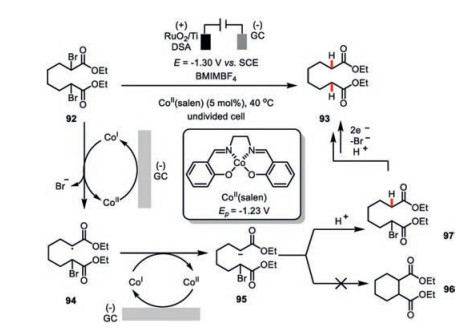
Dehalogenation of the organohalides would be the dominant reaction pathway if there is lack of the π-bond acceptor available in the electrochemical cobalt catalysis. Fuchigami and co-workers has demonstrated the Co(salen) as an effective catalyst for the electrochemical debromination of compound 92 (Scheme 16) in 1-butyl-3-methylimidazolium tetrafluoroborate (BMIMBF4) as the ionic liquid. Since the redox potential of Co(Ⅲ/Ⅱ) is -1.23 V vs. SCE, the operating potential only needed to be a slightly more negative, i.e., -1.30 V [58]. Under these electrochemical setting, the catalytically active low-valent Co(Ⅰ) initially undergoes a single-electron transfer with compound 92 to generate an alkyl radical 94, which could be further reduced to the corresponding carbon anion 95. To be noted, only the protonation of 95 instead of an intramolecular cyclization (96) [59] occurred probably due to the viscous feature of the ionic liquid. Therefore, a second cascade debromination/protonation sequence led to the final product 93.

|
Download:
|
| Scheme 16. Cobaltaelectro-catalyzed electrochemical debromination [58]. | |
{kind=link}
3.2. Cobaltaelectro-catalyzed reductive C—C bond formation towards π-acceptors
Scheffold and co-workers disclosed an elegant vitamin B12-catalyzed electrochemical intramolecular 1, 4-addition of alkyl bromides (Scheme 17) [60]. The electrolysis was operated under constant potential in the rage of -1.4 to -1.9 V vs. Ag/Ag+ in a divided cell since the postulated catalytically active Co(Ⅰ) species could be generated via a two-step reduction with E1/2(1) = -0.36 V and E1/2(2) = -1.08 V. Well agreement with the Baldwin's rules, six-and seven-membered rings by endocyclic closure as well as five-and six-membered rings by exocyclic closure were favored. This transformation was believed to proceed an alkyl Co(Ⅲ) intermediate 100, which can be electrochemically prepared and spectroscopically characterized.

|
Download:
|
| Scheme 17. Electrochemical vitamin B12-catalyzed intramolecular 1, 4-addition [60]. | |
{kind=link}
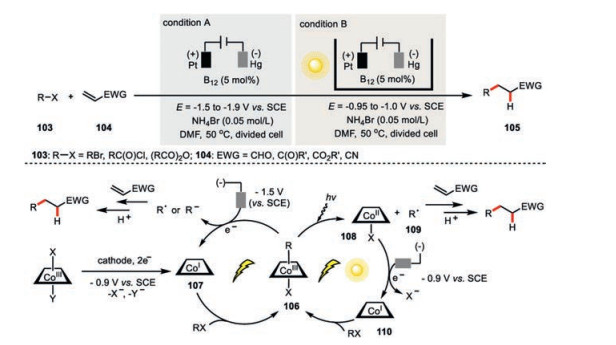
This vitamin B12-catalyzed electrochemical 1, 4-addition reaction could also be well applied intermolecularly to a variety α, β-unsaturated acceptors 104 (including α, β-unsaturated aldehyde, ketone, ester and nitrile) using alkyl bromides, acyl chlorides, and anhydrides (Scheme 18) [61-64]. Interestingly, while electrochemical conditions (condition A) required the operative potential as negative as -1.5 V to -1.9 V, the merger of photochemistry and electrochemistry (condition B) dramatically reduced the necessary potential to be -0.95 to -1.0 V. Since both reaction conditions involve the alkyl Co(Ⅲ) intermediate 106, the different operating potentials originated from the diverted catalytic destiny of such a species. The electrochemical pathway needs to directly reduce the Co(Ⅲ) 106 to Co(Ⅰ) 107 with a reduction potential as negative as -1.5 V. On the contrary, an initial homolysis of the alkyl Co(Ⅲ) species 106 leads to Co(Ⅱ) 108 and an alkyl radical 109 in the photoelectrochemical pathway. The alkyl radical 109 then undergoes further transformations to the desired product. Therefore, the operating potential in this case is only responsible for the reduction of Co(Ⅱ) 108 to Co(Ⅰ) 110 (E = -0.9 V). This is a very elegant example of photoelectrochemistry accelerating the discovery of new reactivity by reducing the innate redox potential of a specific reaction thus much milder reaction conditions and improved functional compatibility could be realized.

|
Download:
|
| Scheme 18. Electrochemical and photoelectrochemical vitamin B12-catalyzed 1, 4-addition [61-64]. | |
{kind=link}
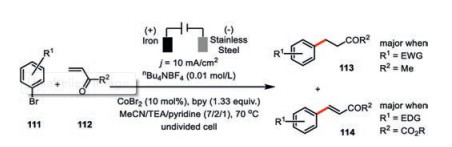
The electrochemical 1, 4-addition of aryl bromides 111 to methyl vinyl ketone 112 was also developed by Gosmini and co-workers using their cobalt-bipyridine system accompanied by a sacrificial iron anode (Scheme 19) [65, 66]. Such catalysis was found to be very sensitive to the choice of substrates. For instance, the major products turned to Heck-type products 114 when acrylates esters were employed. Interestingly, they also observed the correlation between product distribution and the electronic property of substituents at the aryl bromides. The electro-donating group (EDG) at the aryl bromides favored 1, 4-addition 113 while the electron-withdrawing group (EWG) preferred the formation of Heck-type products 114. However, there is still lack of an operating rationale for such a preference.

|
Download:
|
| Scheme 19. Cobaltaelectro-catalyzed electrochemical 1, 4-addition and Heck-type reaction [65, 66]. | |
{kind=link}
Torri and co-workers developed a choloropyridinecobaloxime(Ⅲ)-catalyzed intramolecular radical cyclization of bromo acetals 115 to the unactivated olefins and alkynes (Scheme 20) [67]. One prominent feature of such an electrochemical transformation versus the conventional trialkyltin hydride promoted radical reaction in aprotic medium was its well tolerance of methanol as solvent. The cyclization was well agreement with the Baldwin's rule as only the exo-cyclization to a five-membered ring 116 was formed.

|
Download:
|
| Scheme 20. Cobaltaelectro-catalyzed electrochemical intramolecular cyclization [67]. | |
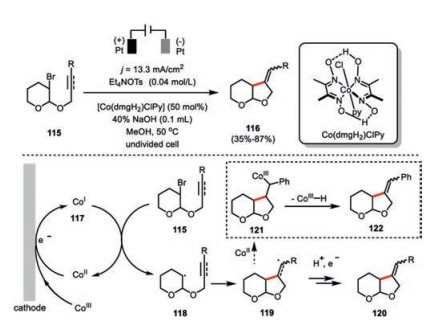{kind=link}
The electrochemically generated Co(Ⅰ) species 117 was proposed to undergo a single-electron transfer with the bromo acetal 115 to deliver the alkyl radical 118, which would proceed a facile 5-exo-dig or 5-exo-trig cyclization to form the furan derivative 120. Interestingly, the alkyl radical 119 generated from the intramolecular cyclization of the phenyl ethylene tethered bromo acetal could be readily intercepted by the Co(Ⅱ) species and then proceed a β-hydride elimination leading to the formation of unsaturated stereoisomer 122. Similar intramolecular reductive cyclization of allyl 2-bromophenyl ether catalyzed by a chiral Jacobson Co(salen) complex only afforded the cyclization product with highest 16% enantiomerical excess [68]. This observation suggested that achieving the desired highly asymmetric cyclization should probably require the identification of a system with a more intimate role of the chiral cobalt complex in the enantio-determining step within the catalysis.
3.3. Cobaltaelectro-catalyzed carbon dioxide fixationThe low-valent Co(Ⅰ) species 122, readily obtained by the reduction of Co(salen) with sodium, was demonstrated by Floriani and co-workers to be capable to absorb an equal molar of CO2 in a highly reversible manner (Scheme 21) [69]. Though the CO2 adduct 123 quickly released CO2 at room temperature in vacuo, the axial pyridine ligation of complex 124 profoundly enhanced its stability. The IR spectra analysis confirmed the incorporation of CO2 to these cobalt complexes. Mechanistically, CO2 was proposed to be activated by the sodium cation as a Lewis acid, which is readily poised to be nucleophilic attached by the super-nucleophilic low-valent Co(Ⅰ) species 122. Following this significant unveiling of low-valent cobalt species towards the activation of CO2, diverse cobalt-catalyzed electrochemical carboxylation of CO2 with activated benzylic/allylic chlorides [70, 71] and chloroacetonitrile [72] were then developed.

|
Download:
|
| Scheme 21. Reversible CO2 fixation of the chemically generated low-valent cobalt(Ⅰ) species [69]. | |
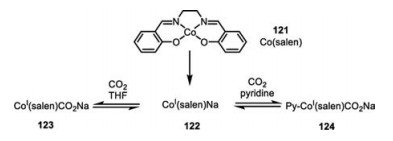{kind=link}
The cobalt-catalyzed chemical carboxylation using carbon dioxide frequently requires the use of excess amount of metal reductant such as zinc, manganese, or Et2Zn [73]. Therefore, the pervasive benzyl and allylic halides are excluded as the potential substrates as they are prone to unproductively react with these reducing agents prior to the carboxylation. By contrast, the cobaltaelectrocatalysis brings forth new reactivity to these activated alkyl halides.
For instance, by using an enantiomerically pure Jacobsen Co(salen) complex, an asymmetric electrocarboxylation of 1-phenylethyl chloride (126, with ee up to 83%) was developed by Wang, Lu, and their co-workers (Scheme 22) [74]. Cyclic voltammetry studies indicated the reduction potential of CoII(salen) 127 to CoI(salen) 128 to be -1.5 V vs. SCE. Being a supernucleophile, this electrochemically generated cobalt(Ⅰ) then undergoes an oxidative addition with the 1-phenylethyl chloride 129 (E=2.19 V vs. SCE) to give a cobalt(Ⅲ) species 130. An additional electron-transfer event delivers the cobalt(Ⅲ) 130 to cobalt(Ⅱ) 131, which bears competent capacity to nucleophilic attack towards CO2 and thus transfers the chiral information from the cobalt complex to the carboxylation product 132. Entrapment of this chiral cobalt complex with silver provided a novel stable and recyclable heterogenous catalyst for the enantioselective carboxylation of benzyl bromides (ee up to 67%) [75].

|
Download:
|
| Scheme 22. Cobaltaelectro-catalyzed electrochemical asymmetric carboxylation [74]. | |
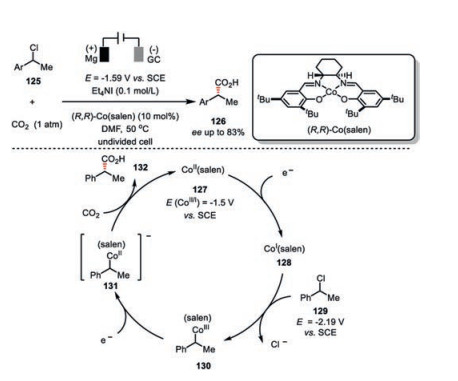{kind=link}
Recently, Ackermann and co-workers found the electrochemically generated low-valent cobalt species 135 from the combination of Co(OAc)2 and PPh3 exhibited much higher activity than previously identified Co(salen) system in the carboxylation of allylic chlorides 133 (Scheme 23) [76]. Detailed cyclic voltammetry studies supported such an electrochemically generated cobalt(Ⅰ) species 135 should readily insert into the allylic C-Cl bond via an oxidative addition, giving rise to the η3-allyl-cobalt(Ⅲ) complex 136. According to their DFT calculations, the isomerization barrier from η3-allyl-cobalt(Ⅲ) complex 136 to η1-allyl-cobalt(Ⅲ) complex 137 or 138 is only 16.1 kcal/mol and thus such an event should not be the rate-determining step. The rearrangement regioselectivity of these allyl-cobalt(Ⅲ) complexes and subsequent electrochemical reduction to their allyl-cobalt(Ⅰ) complexes 139 or 140, in terms of the position of cobalt, is highly depending on the coordination environment. The low-valent cobalt complexes then proceed a γ-selective SN2′ carboxylation towards CO2 resulting in 2-phenylbut-3-enoic acid derivative 141 and styrylacetic acid derivative 142 in moderate regioselectivity. By contrast to the previously mechanistic proposal where cobalt(Ⅱ)-salen 131 (Scheme 22) is responsible for the carboxylation, cobalt(Ⅰ) species (139 and 140) is more likely to exist based on their mechanistic investigation, and then proceed carboxylation towards CO2 under current reaction setting. These examples again highlight the crucial role of ligand in the cobaltaelectrocatalysis.

|
Download:
|
| Scheme 23. Cobaltaelectro-catalyzed carboxylation of allylic chlorides [76]. | |
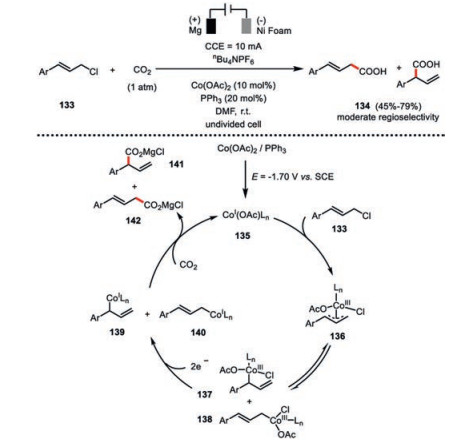{kind=link}
4. Conclusion
In summary, recent advances in the merger of cobalt catalysis and electrochemistry in organic synthesis including the oxidative and reductive cobaltaelectrocatalysis are highlighted. The introduction of electrochemistry further enriches the versatile redox reactivity of cobalt catalysis and thus helps to profoundly expand the synthetic boundary. Such a constructive merger makes the cobaltaelectrocatalysis even greener and more sustainable as electricity is incorporated as the traceless oxidants and reductants.
Being a very promising strategy in the organic synthesis, the merging of cobalt catalysis and electrochemistry also meets some limitations. For instance, most of the applicable reaction types are limited to the known processes using chemical oxidants/reductants. In addition, the investigations of asymmetric variants in the cobaltaelectrocatalysis are quite rare despite the state-of-the-art of cobalt in the asymmetric catalysis. Therefore, future directions of the investigation of merging cobalt catalysis and electrochemistry should aim to solve current limitations. The innovative incorporation of cobalt catalysis in electrochemistry as well as photoelectrochemistry [77-82] has profoundly expanded the current synthetic boundary of cobalt and should be very promising means for the searching of novel reactivity as well as high selectivity.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (No. 21901041) and Fuzhou University (No. 510841) is gratefully acknowledged.
| [1] |
M. Hapke, G. Hilt, Cobalt Catalysis in Organic Synthesis: Methods and Reactions, 1st. ed., Wiley, Weinheim, 2020.
|
| [2] |
C.K. Rofer-DePoorter, Chem. Rev. 81 (1981) 447-474. DOI:10.1021/cr00045a002 |
| [3] |
H. Schulz, Appl. Catal. A: Gen. 186 (1999) 3-12. DOI:10.1016/S0926-860X(99)00160-X |
| [4] |
K.L. Brown, Chem. Rev. 105 (2005) 2075-2149. DOI:10.1021/cr030720z |
| [5] |
M. Giedyk, K. Goliszewska, D. Gryko, Chem. Soc. Rev. 44 (2015) 3391-3404. DOI:10.1039/C5CS00165J |
| [6] |
J. Cheng, L. Wang, P. Wang, L. Deng, Chem. Rev. 118 (2018) 9930-9987. DOI:10.1021/acs.chemrev.8b00096 |
| [7] |
Y. Liu, J. Cheng, L. Deng, Acc. Chem. Res. 53 (2019) 244-254. |
| [8] |
K. Gao, N. Yoshikai, Acc. Chem. Res. 47 (2014) 1208-1219. DOI:10.1021/ar400270x |
| [9] |
T. Michiyuki, K. Komeyama, Asian J. Org. Chem. 9 (2019) 343-358. |
| [10] |
S. Mohle, M. Zirbes, E. Rodrigo, et al., Angew. Chem. Int. Ed. 57 (2018) 6018-6041. DOI:10.1002/anie.201712732 |
| [11] |
A. Wiebe, T. Gieshoff, S. Mohle, et al., Angew. Chem. Int. Ed. 57 (2018) 5594-5619. DOI:10.1002/anie.201711060 |
| [12] |
S. Tang, Y. Liu, A. Lei, Chem 4 (2018) 27-45. DOI:10.1016/j.chempr.2017.10.001 |
| [13] |
J. Yoshida, K. Kataoka, R. Horcajada, A. Nagaki, Chem. Rev. 108 (2008) 2265-2299. DOI:10.1021/cr0680843 |
| [14] |
Z. Li, Q. Sun, P. Qian, et al., Chin. Chem. Lett. 31 (2020) 1855-1858. DOI:10.1016/j.cclet.2020.02.030 |
| [15] |
J. Zhang, H. Wang, Y. Chen, et al., Chin. Chem. Lett. 31 (2020) 1576-1579. DOI:10.1016/j.cclet.2019.11.037 |
| [16] |
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230-13319. DOI:10.1021/acs.chemrev.7b00397 |
| [17] |
R. Francke, R.D. Little, Chem. Soc. Rev. 43 (2014) 2492-2521. DOI:10.1039/c3cs60464k |
| [18] |
K.J. Jiao, Y.K. Xing, Q.L. Yang, H. Qiu, T.S. Mei, Acc. Chem. Res. 53 (2020) 300-310. DOI:10.1021/acs.accounts.9b00603 |
| [19] |
J.C. Siu, N. Fu, S. Lin, Acc. Chem. Res. 53 (2020) 547-560. DOI:10.1021/acs.accounts.9b00529 |
| [20] |
P. Gandeepan, L.H. Finger, T.H. Meyer, L. Ackermann, Chem. Soc. Rev. 49 (2020) 4254-4272. DOI:10.1039/D0CS00149J |
| [21] |
T.H. Meyer, L.H. Finger, P. Gandeepan, L. Ackermann, Trends Analyt. Chem. 1 (2019) 63-76. DOI:10.1016/j.trechm.2019.01.011 |
| [22] |
Y. Jiang, K. Xu, C. Zeng, Chem. Rev. 118 (2018) 4485-4540. DOI:10.1021/acs.chemrev.7b00271 |
| [23] |
P. Xiong, H.C. Xu, Acc. Chem. Res. 52 (2019) 3339-3350. DOI:10.1021/acs.accounts.9b00472 |
| [24] |
N. Sauermann, T.H. Meyer, C. Tian, L. Ackermann, J. Am. Chem. Soc. 139 (2017) 18452-18455. DOI:10.1021/jacs.7b11025 |
| [25] |
X.R. Chen, S.Q. Zhang, T.H. Meyer, et al., Chem. Sci. 11 (2020) 5790-5796. DOI:10.1039/D0SC01898H |
| [26] |
T.H. Meyer, J.C.A. Oliveira, D. Ghorai, L. Ackermann, Angew. Chem. Int. Ed. 59 (2020) 10955-10960. DOI:10.1002/anie.202002258 |
| [27] |
L. Ackermann, Acc. Chem. Res. 53 (2020) 84-104. DOI:10.1021/acs.accounts.9b00510 |
| [28] |
N. Sauermann, T.H. Meyer, L. Ackermann, Chem. Eur. J. 24 (2018) 16209-16217. DOI:10.1002/chem.201802706 |
| [29] |
C. Tian, U. Dhawa, J. Struwe, L. Ackermann, Chin. J. Chem. 37 (2019) 552-556. DOI:10.1002/cjoc.201900050 |
| [30] |
X. Gao, P. Wang, L. Zeng, S. Tang, A. Lei, J. Am. Chem. Soc. 140 (2018) 4195-4199. DOI:10.1021/jacs.7b13049 |
| [31] |
N. Sauermann, R. Mei, L. Ackermann, Angew. Chem. Int. Ed. 57 (2018) 5090-5094. DOI:10.1002/anie.201802206 |
| [32] |
U. Dhawa, C. Tian, W. Li, L. Ackermann, ACS Catal. 10 (2020) 6457-6462. DOI:10.1021/acscatal.0c01436 |
| [33] |
T.H. Meyer, G.A. Chesnokov, L. Ackermann, ChemSusChem 13 (2020) 668-671. DOI:10.1002/cssc.202000057 |
| [34] |
C. Tian, L. Massignan, T.H. Meyer, L. Ackermann, Angew. Chem. Int. Ed. 57 (2018) 2383-2387. DOI:10.1002/anie.201712647 |
| [35] |
Y. Cao, Y. Yuan, Y. Lin, et al., Green Chem. 22 (2020) 1548-1552. DOI:10.1039/D0GC00289E |
| [36] |
R. Mei, N. Sauermann, J.C.A. Oliveira, L. Ackermann, J. Am. Chem. Soc. 140 (2018) 7913-7921. DOI:10.1021/jacs.8b03521 |
| [37] |
R. Mei, X. Fang, L. He, et al., Chem. Commun. 56 (2020) 1393-1396. DOI:10.1039/C9CC09076B |
| [38] |
T.H. Meyer, J.C.A. Oliveira, S.C. Sau, N.W.J. Ang, L. Ackermann, ACS Catal. 8 (2018) 9140-9147. DOI:10.1021/acscatal.8b03066 |
| [39] |
S. Tang, D. Wang, Y. Liu, L. Zeng, A. Lei, Nat. Commun. 9 (2018) 798. DOI:10.1038/s41467-018-03246-4 |
| [40] |
L. Zeng, H. Li, S. Tang, et al., ACS Catal. 8 (2018) 5448-5453. DOI:10.1021/acscatal.8b00683 |
| [41] |
J. Chen, L. Jin, J. Zhou, X. Jiang, C. Yu, Tetrahedron Lett. 60 (2019) 2054-2058. DOI:10.1016/j.tetlet.2019.06.060 |
| [42] |
S.C. Sau, R. Mei, J. Struwe, L. Ackermann, ChemSusChem 12 (2019) 3023-3027. DOI:10.1002/cssc.201900378 |
| [43] |
Y.L. Lai, J.S. Ye, J.M. Huang, Chem. Eur. J. 22 (2016) 5425-5429. DOI:10.1002/chem.201505074 |
| [44] |
Y. Yu, P. Zheng, Y. Wu, X. Ye, Org. Biomol. Chem. 16 (2018) 8917-8921. DOI:10.1039/C8OB02485E |
| [45] |
S. Tian, S. Lv, X. Jia, et al., Adv. Synth. Catal. 361 (2019) 5626-5633. DOI:10.1002/adsc.201901260 |
| [46] |
S. Lin, R.A. DiStasio Jr., M.O. Frederick, et al., Nat. Chem. (2020). DOI:10.1038/s41557-020-0469-5 |
| [47] |
J.M. Hammann, M.S. Hofmayer, F.H. Lutter, L. Thomas, P. Knochel, Synthesis 49 (2017) 3887-3894. DOI:10.1055/s-0036-1588430 |
| [48] |
T. Fujihara, Y. Tsuji, Beilstein J. Org. Chem. 14 (2018) 2435-2460. DOI:10.3762/bjoc.14.221 |
| [49] |
E. Le Gall, C. Gosmini, J.Y. Nédélec, J. Périchon, Tetrahedron Lett. 42 (2001) 267-269. DOI:10.1016/S0040-4039(00)01936-5 |
| [50] |
P. Gomes, H. Fillon, C. Gosmini, E. Labbé, J. Périchon, Tetrahedron 58 (2002) 8417-8424. DOI:10.1016/S0040-4020(02)01030-X |
| [51] |
P. Gomes, C. Gosmini, J. Périchon, Tetrahedron 59 (2003) 2999-3002. DOI:10.1016/S0040-4020(03)00404-6 |
| [52] |
L. Polleux, E. Labbé, O. Buriez, J. Périchon, Chem. Eur. J. 11 (2005) 4678-4686. DOI:10.1002/chem.200400971 |
| [53] |
P. Gomes, C. Gosmini, J. Périchon, Org. Lett. 5 (2003) 1043-1045. DOI:10.1021/ol0340641 |
| [54] |
P. Gomes, C. Gosmini, J. Périchon, J. Org. Chem. 68 (2003) 1142-1145. DOI:10.1021/jo026421b |
| [55] |
C. Gosmini, Y. Rollin, J.Y. Nédélec, J. Périchon, J. Org. Chem. 65 (2000) 6024-6026. DOI:10.1021/jo000459b |
| [56] |
E.L. Gall, C. Gosmini, Y. J.-Nédélec, J. Périchon, Tetrahedron 57 (2001) 1923-1927. DOI:10.1016/S0040-4020(01)00015-1 |
| [57] |
H. Fillon, C. Gosmini, J. Périchon, Tetrahedron 59 (2003) 8199-8202. DOI:10.1016/j.tet.2003.08.032 |
| [58] |
Y. Shen, S. Inagi, M. Atobe, T. Fuchigami, Res. Chem. Intermed. 39 (2013) 89-99. DOI:10.1007/s11164-012-0634-6 |
| [59] |
M. Tokuda, A. Hayashi, H. Suginome, Bull. Chem. Soc. Jpn. 64 (1991) 2590-2592. DOI:10.1246/bcsj.64.2590 |
| [60] |
R. Scheffold, M. Dike, S. Dike, T. Herold, L. Walder, J. Am. Chem. Soc. 102 (1980) 3642-3644. DOI:10.1021/ja00530a064 |
| [61] |
L. Auer, C. Weymuth, R. Scheffold, Helv. Chim. Acta 76 (1993) 810-818. DOI:10.1002/hlca.19930760205 |
| [62] |
R. Orlinski, T. Stankiewicz, Tetrahedron Lett. 29 (1988) 1601-1602. DOI:10.1016/S0040-4039(00)80364-0 |
| [63] |
R. Scheffold, R. Orlinski, J. Am. Chem. Soc. 105 (1983) 7200-7202. DOI:10.1021/ja00362a047 |
| [64] |
L. Walder, G. Rytz, K. Meier, R. Scheffold, Helv. Chim. Acta 61 (1978) 3013-3017. DOI:10.1002/hlca.19780610826 |
| [65] |
P. Gomes, C. Gosmini, J.Y. Nédélec, J. Périchon, Tetrahedron Lett. 41 (2000) 3385-3388. DOI:10.1016/S0040-4039(00)00420-2 |
| [66] |
P. Gomes, C. Gosmini, J.Y. Nédélec, J. Périchon, Tetrahedron Lett. 43 (2002) 5901-5903. DOI:10.1016/S0040-4039(02)01286-8 |
| [67] |
S. Torii, T. Inokuchi, T. Yukawa, J. Org. Chem. 50 (1985) 5875-5877. DOI:10.1021/jo00350a090 |
| [68] |
E. Duñach, A.P. Esteves, M.J. Medeiros, D. Pletcher, S. Olivero, J. Electroanal. Chem. 566 (2004) 39-45. DOI:10.1016/j.jelechem.2003.10.045 |
| [69] |
C. Floriani, G. Fachinetti, J. Chem. Soc., Chem. Commun. (1974) 615-616. |
| [70] |
J.C. Folest, J.M. Duprilot, J. Perichon, Y. Robin, J. Devynck, Tetrahedron Lett. 26 (1985) 2633-2636. DOI:10.1016/S0040-4039(00)98122-X |
| [71] |
G. Zheng, M. Stradiotto, L. Li, J. Electroanal. Chem. 453 (1998) 79-88. DOI:10.1016/S0022-0728(98)00173-9 |
| [72] |
L. P.-Fabre, O. Reynes, Electrochem. Commun. 12 (2010) 1360-1362. DOI:10.1016/j.elecom.2010.07.020 |
| [73] |
D.J. Weix, Acc. Chem. Res. 48 (2015) 1767-1775. DOI:10.1021/acs.accounts.5b00057 |
| [74] |
L. B.-Chen, W. H.-Zhu, Y. Xiao, et al., Electrochem. Commun. 42 (2014) 55-59. DOI:10.1016/j.elecom.2014.02.009 |
| [75] |
H.P. Yang, Y.N. Yue, Q.L. Sun, et al., Chem. Commun. 51 (2015) 12216-12219. DOI:10.1039/C5CC04554A |
| [76] |
N.W.J. Ang, J.C.A. Oliveira, Ackermann L., Angew. Chem. Int. Ed. (2020). DOI:10.1002/anie.202003218 |
| [77] |
L. Capaldo, L.L. Quadri, D. Ravelli, Angew. Chem. Int. Ed. 58 (2019) 17508-17510. DOI:10.1002/anie.201910348 |
| [78] |
Y. Yu, P. Guo, J.S. Zhong, Y. Yuan, K.Y. Ye, Org. Chem. Front. 7 (2020) 131-135. DOI:10.1039/C9QO01193E |
| [79] |
J.P. Barham, B. König, Angew. Chem. Int. Ed. 59 (2020) 11732-11747. DOI:10.1002/anie.201913767 |
| [80] |
T. Hardwick, A. Qurashi, B. Shirinfar, N. Ahmed, ChemSusChem 13 (2020) 1967-1973. DOI:10.1002/cssc.202000032 |
| [81] |
Y.C. Wu, R.J. Song, J.H. Li, Org. Chem. Front. 7 (2020) 1895-1902. DOI:10.1039/D0QO00486C |
| [82] |
J. Liu, L. Lu, D. Wood, S. Lin, ACS Cent. Sci. (2020). DOI:10.1021/acscentsci.0c00549 |