 2021, Vol. 32
2021, Vol. 32 


b School of Chemical and Material Science, Key Laboratory of Magnetic Molecules & Magnetic Information Materials, Ministry of Education, Shanxi Normal University, Linfen 041004, China;
c University of Chinese Academy of Sciences, Beijing 100049, China
The low-temperature oxidation of CO has received considerable attention from the environmental and material scientists, due to its important role in controlling of vehicle exhaust emissions and purification of gas streams derived from petrochemical industry [1-3]. Particularly interesting are the nanoscale catalysts, the composition and structure of which can be tailored to fulfill the desired chemical behaviors [4-6]. With the aid of state-of-art in- situ spectrum technologies, two dynamically distinct types of bimolecular surface reactions have been proposed in the heterogeneous catalysis of CO oxidation, which are now denoted as Langmuir–Hinshelwood (LH)- and Eley–Rideal (ER)-type mechanism, respectively [7]. In the LH mechanism, both the CO molecule and oxygen species are coadsorpted on the catalyst surface, and subsequently, the CO2 is formed by either the rearrangement between chemisorbed CO and oxygen, or direct attachment of the CO ligand to the near-neighbor oxygen centers (intrasystem attack) [8]. On the contrary, in the ER mechanism, the CO molecule from the gas phase reacts directly with the chemisorbed oxygen species on the catalyst surface to give rise to CO2 (intersystem attack) [8].
Microscopically, the real-life catalytic event usually takes place on a specific active site consisting of only a small number of atoms, of which the particular electronic, geometric, and bonding properties are the root of selectivity of CO oxidation mechanism. The gas-phased cluster reaction researches performed under isolated, size-controllable, and reproducible conditions provide an alternative route to capture clear molecular- and electronic-level mechanisms of the catalytic CO oxidation. The CO oxidation of the binary transition metal oxide (TMO) clusters serves as a well-defined model pertinent to mechanistic understandings of the surface catalysis. Recent researches of particular interest are the heteronuclear TMO clusters and highlight the potential to modulate chemical processes by selective cluster doping [9, 10]. The different metallic fractions of heteronuclear TMO clusterscan mimic either the individual active sites or their supports of real-life catalysts [11]. The well similar behaviors paralleled with the condensed-phase CO oxidation by supported catalysts have been found in the gas-phase catalytic CO oxidation mediated by nanocatalysts doped by noble-metal single-atom and dimer [12-15]. The noble-metal-like behavior in many catalytic processes has also been found for a few noble-metal-free heteronuclear TMO clusters [14-16]. The high adsorption energy of CO on the atomic or dimeric dopants and dynamic nature of dopants in terms of the electron storage and release are found to be the driving force for the CO oxidation [17]. Due to the strong carbonyl binding to the doped TM, especially for the noble TM, the atomic TM dopant usually functions as a preferred trapping site and electron acceptor for CO adsorption and then acts as a deliverer of CO ligand for CO oxidation by the oxygen species on the heteronuclear TMO cluster. In this sense, available gas-phase experiments in the literature indicate that the CO oxidation reaction on heteronuclear TMO clusters preferentially proceeds via LH-like mechanism.
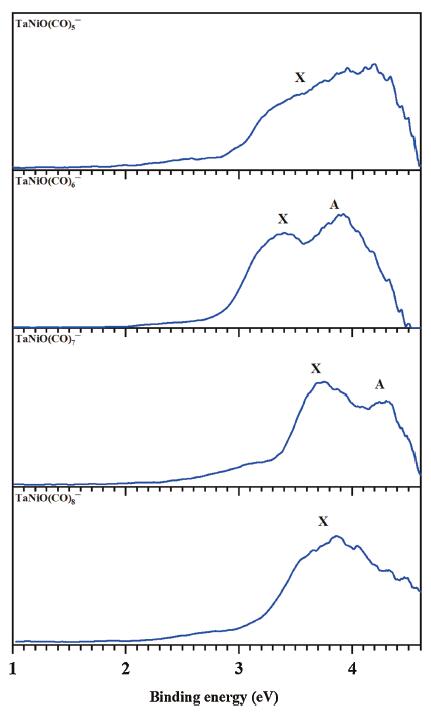
The recently reported literatures have successfully identified ligand-mediated reactivity in the CO oxidation of V-doped [16] and Nb-doped [18] nickel oxide complexes, on which the CO oxidation can occur by LH-like mechanism. Inspired by these facts, we extend our research to their tantalum congeners TaNiO(CO)n− (n=5–8). The details of experiment and computation can be found in the Supporting information.Fig. 1 illustrates the photoelectron spectra of the TaNiO(CO)n− (n=5–8) series obtained at 266 nm. A tabulated summary of the experimentally measured data is given in Table S2 (Supporting information), where they are compared with the theoretically predicted value at the BP86-D3BJ/def2-TZVP level. The spectral signatures are distinguishable from each other for the TaNiO(CO)n− (n=5–8) series, despite the similarly broad bands. One broad band without discernible feature is observed in the spectrum of TaNiO(CO)5−, suggesting a lager structural change upon photodetachment for this anion. The main band in the spectrum of TaNiO(CO)6− is obviously split into two peaks located at 3.42 and 3.91 eV, with a slightly higher intensity in the second peak. The spectrum of TaNiO(CO)7− is split into two peaks located at 3.74 and 4.29 eV, with the first peak becoming dominant. Moreover, the ground-state VDE1 of TaNiO(CO)7− is blue-shifted by over 0.3 eV, as compared to those of TaNiO(CO)5− and TaNiO(CO)6−. Such appreciable differences in the spectral feature imply that TaNiO(CO)7− should have different geometry from TaNiO(CO)n− (n=5–6), as validated by the undermentioned theoretical calculations. However, only one broad band with the maximum located at 3.86 eV is revealed in the spectrum of TaNiO(CO)8−. Overall, the similarities and differences revealed in the experimental spectra, together with band shift, imply the emergence of geometric evolution during the consecutive CO adsorption on tantalum–nickel monoxide complexes.

|
Download:
|
| Fig. 1. Photoelectron spectra of TaNiO(CO)n− (n = 5–8) recorded at 266 nm (4.661 eV). | |
{kind=link}
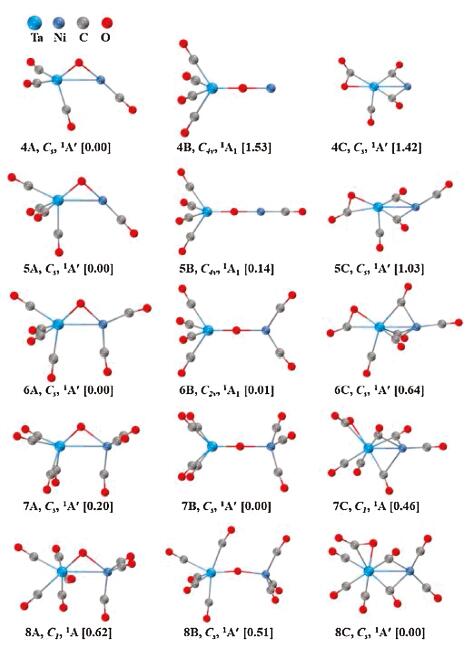
Quantum chemical calculations were carried out to elucidate the geometric and electronic properties of the TaNiO(CO)n− (n=4–8) series. The ground state candidates and alternative structures optimized at the BP86-D3BJ/def2-TZVP level are all closed-shell species and depicted in Fig. S1 (Supporting information). As present in Fig. 2, three kinds of representative geometries with distinct structural characteristics are predicted to participate in the competition of the possible ground states during the continuous CO adsorption progress. The first kind structure in the form of (CO)xTa−(μ2-O)−Ni(CO)n−x is labeled with nA and comprises of a triangular TaONi− anion core and terminal CO ligands bonded to metal atoms. The oxygen atom in the TaONi− core occupies the bridging position between Ta and Ni atoms. We define this type of geometry as a μ2-O-bent structure. The transformation of TaONi− anion core from triangular to linear will create the second typed structure in the form of (CO)xTa−(μ2-O)−Ni(CO)n−x. This kind of structure is defined as μ2-O-linear structure, which involves a linear TaONi− anion core terminally bonded by multiple CO ligands. The third type geometry labeled with nC has a bent CO2 ligand bonded to the Ta center in a side-on η2-C, O fashion, besides n–1 CO ligands either terminally or bridged bonded to the metal atoms. We define this type of structure as a η2-CO2-tagged structure.

|
Download:
|
| Fig. 2. Ground-state structures and selected low-lying isomers of the TaNiO(CO)n− (n = 4–8) anions calculated at the BP86-D3BJ/def2-TZVP level. Relative energies are given in square brackets (eV). | |
{kind=link}
It can be seen from Fig. 2 that these three types of structures energetically compete with each other during the consecutive CO adsorption on tantalum–nickel monoxide complexes. For TaNiO(CO)4−, the most stable isomer is the μ2-O-bent structure with triangle TaONi core (4A). Both the μ2-O-linear isomer (4B) involving linear Ta-O-Ni center and the η2-CO2-tagged isomer (4C) containing a CO2 ligand side-on bonded to Ta atomare thermodynamically unfavorable, which are 1.53 and 1.42 eV higher in energy than 4A, respectively. The large energy spacings hint at the overpowering stability of the μ2-O-bent isomer. Additional attachment of CO ligands to the metal center successively generates higher-coordinated TaNiO(CO)n− (n=5–8) complexes with similar geometric characteristics. However, multiple adsorptions of CO ligands can vary both the relative stability and the energy separations among these three types of isomers. The energy spacing between μ2-O-bent structure and μ2-O-linear structure dramatically decreases when one CO terminally attaches to TaNiO(CO)4−. For TaNiO(CO)5−, the ground-state isomer is predicted to be a μ2-O-bent structure (5A), followed by μ2-O-linear isomer (5B) and η2-CO2-tagged isomer (5C) being 0.14 and 1.03 eV higher in energy, respectively. Additional CO adsorption leads to a further decrease in energy separation and results in two near degenerate isomers 6A and 6B, with isomer 6B being slightly higher in energy. Therefore, the global minimums for TaNiO(CO)n− (n=4–6) are all μ2-O-bent structures, as similar with the congeneric NbNiO(CO)n− (n=4–6) series [18]. However, an inversion of the relative stability of the μ2-O-bent structure and μ2-O-linear structure is achieved when one CO ligand attaches to TaNiO(CO)6−. For n=7, the thermodynamically most stable structure is the μ2-O-linear structure (7B), which is followed sequentially by μ2-O-linear isomer (7A) and η2-CO2-tagged isomer (7C). The energy difference between 7B and 7A/7C is 0.20/0.46 eV. At this point, the situation for TaNiO(CO)7− anion is different. For previously reported congener NbNiO(CO)7− anion, it has already been transformed into the η2-CO2-tagged structure [18]. Such an alteration in the structure occurs again when one more CO ligand is adsorbed onto TaNiO(CO)7−. The relative energetics of the isomers suggest that the ground state of TaNiO(CO)8− is the most competitive isomer 8C, i.e., η2-CO2-tagged structure, which is 0.62 and 0.51 eV lower in energy than the μ2-O-bent isomer (8A) and μ2-O-linear structure (8B), respectively. In conclusion, during the consecutive CO adsorption, the μ2-O-bent structure theoretically is the most favorable for TaNiO(CO)n− (n=4–6), then the μ2-O-linear isomer is more preferential for TaNiO(CO)7−, and finally the η2-CO2-tagged geometry is predicted to be the most energetically competitive one for TaNiO(CO)8−.
Table S2 provides a comparison of the theoretical calculated VDEs and ADEs at the BP86-D3BJ/def2-TZVP level and the experimental data. Considerable reorganization energies (ROEs) are predicted for all of the complexes, correlating well with the experimentally observed broad band in the PES. The ROE is defined as the energy difference between the ground-state ADE and VDE, the amount of which roughly characterizes the anion-to-neutral structural relaxation upon electron detachment. As an aside, it is interesting to note that the predicted VDEs for the nA isomers with a triangular TaONi− anion core and nC isomers involving a bent CO2 ligand are approximately located at around 3.4 and 3.7 eV, respectively. While for the μ2-O-linear structure, the VDE roughly increases with the multipleadsorptions of CO ligands.
For the PES of TaNiO(CO)n− (n=5–7) anions, the intensity of η2-CO2-tagged isomer 5C, 6C and 7C are anticipated to be thermodynamically negligible and therefore can be excluded, due to their overestimated ADEs and VDEs and high relative energy of 1.03, 0.64 and 0.46 eV. Moreover, as can be seen from Fig. S2 (Supporting information), their simulated spectra disagree with the experimental PES. The major competition occurs between the μ2-O-bent structure and μ2-O-linear structure. The ground-state structure of TaNiO(CO)5− is μ2-O-bent isomer 5A, of which the predicted ADE and VDE are 3.01 and 3.45 eV, respectively, in accordance with experimental values. The ADE and VDE of isomer 5B are estimated to be 3.17 and 3.27 eV, respectively. The comparatively small ROE suggests that the μ2-O-linear structure is relatively rigid. The situation is slightly more complicated for TaNiO(CO)6−. The μ2-O-bent isomer 6A and μ2-O-linear isomer 6B are predicted to be energetically near-degenerate, with 6A locating slightly lower in energy. The ground-state ADE and VDE of isomer 6A are 3.10 and 3.41 eV, respectively, in good agreement with the experiment. The theoretical first excited state VDE2 (4.11 eV) is in line with the second peak observed in the PES. The VDE of isomer 6B is predicted to be 3.64 eV, which is a little bit blue-shifted with respect to the experimental data. For TaNiO(CO)7−, only the ground-state ADE and VDEs of isomer 7B are excellently consistent with the experimentally measured values. Moreover, the predicted VDE (4.26 eV) of the first excited state for isomer 7B excellently agrees with the second peak in the experimental PES. However, the ADE and VDE of isomer 7A are underestimated with respect to the experiment, while the ones for isomer 7C are overestimated. For TaNiO(CO)8−, the experimental PES can be assigned to the structure 8C, for which the predicted ADE and VDE are in good agreement with the experimental data. Alternative isomers, such as μ2-O-bent isomer 8A and μ2-O-linear isomer 8B, are thermodynamically unfavorable and therefore excludable. Furthermore, the VDE of isomer 8A is severely underestimated relative to the experimental value, while the ADE of isomer 8B is overestimated.
Additionally, the spectra have been simulated based on the theoretically modeled excitation energies of the three types of isomers using time-dependent density functional theory (TDDFT). The simulated spectra are depicted in Fig. S2 and compared with the experimental PES. For the competitive isomers 5A and 5B, only the simulated PES of isomer 5A matches the experimental one. The fit PES of 5B has two separate peaks, which disagrees with the experimental PES. Thus, the observed PES is derived from the photodetachment of isomer 5A anion. For TaNiO(CO)6−, both of competitive isomers 6A and 6B have two partially overlapping peaks. As compared with isomer 6B, the first peak of isomer 6A fit more with the observed peak X, while the secondary peak is blue-shifted with respect to the experimental peak A. Moreover, the ratio of the simulated two peaks mismatches the experimental PES. For the case of 6B, both two peaks are blue-shifted with respect to the experimental peaks. However, despite that, the ratio of the fitted two peaks agrees with the experiment. In this sense, both of the near-degenerate isomers potentially coexist and contribute to the experimental spectrum. Similarly, two separate peaks are modeled for both isomers 7A and 7B. However, only the spectral pattern of isomer 7B is in accordance with experimental PES of TaNiO(CO)7−, while the second of two peaks is dominant for isomer 7A, which deviates from the observed spectrum. Thus, both the predicted ADE and VDE and simulated PES suggest μ2-O-linear structure 7B as the ground-state structure of TaNiO(CO)7−. For TaNiO(CO)8−, only the simulated spectrum of ground-state 8C reasonably coincides with experimental observation. While for the isomers 8A and 8B, the simulated spectrum has either two separate peaks or only one deviated peak, which is found to be incompatible with the experiment. Therefore, the observed PES belongs to the η2-CO2-tagged isomer 8C. Comprehensive comparison between the experiment and theory coincidentally demonstrate that, for the consecutive CO adsorption process of the TaNiO(CO)n− (n=5–8) series, the μ2-O-bent structure is initially preferential, then the μ2-O-linear structure is thermodynamically favorable, and finally, the η2-CO2-tagged structure becomes dominant. The details of comparison can be found in the electronic supplementary information. It is demonstrated that structural transformation occurs during the successive CO adsorption, and the CO oxidation happens at n=8.
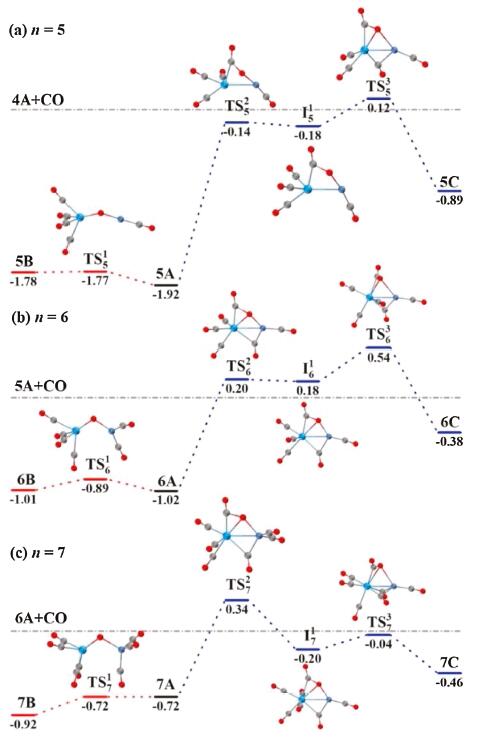
To rationalize the experimental results and provide insights into the dynamic reactivity of TaNiO(CO)n− complexes toward CO oxidation, the structural isomerizations among three types of isomers were theoretically analyzed at the BP86-D3BJ/def2-TZVP level. For TaNiO(CO)n− (n=5–7), the potential energy profiles of the structural isomerizationsare shown in Fig. 3. The left part of Fig. 3 (shown in red) are potential energy profiles of the isomerizations between the μ2-O-bent isomer and the μ2-O-linear isomer. The isomerization mainly involves a geometrical transformation of the TaONi core, namely, the variation of TaONi bond angle or Ta-Ni bond length. The low energy barriers for the isomerization step correlate well with the small Mayer bond orders of Ta-Ni bond summarized in Table S3 (Supporting information). There is almost no barrier for the isomerization reaction from isomer 5B to the more stable isomer 5A. Whereas, there is no barrier for the inverse reaction at n=7, i.e., the isomerization from isomer 7A to the more stableisomer 7B. These explain the absence of isomer 5B and isomer 7A in the PES of TaNiO(CO)5− and TaNiO(CO)7−, respectively. For n=6, however, the isomerization reaction is almost thermoneutral between the degenerate isomers 6A and 6B, which are separated by the transition state with a barrier of 0.13 eV. Both of them may present in the anionic beam of TaNiO(CO)6−. It is demonstrated that the presence of μ2-O-bent isomer and/or μ2-O-linear isomer is a consequence of quantitatively different reaction barriers and relative thermodynamic stability between these isomers.

|
Download:
|
| Fig. 3. DFT calculated potential energy profiles for the isomerization reactions (nB → nA → nC) of TaNiO(CO)n− (n = 5–7). The zero-point vibration corrected energies (eV) with respect to the separated reactants (n−1)A and CO are given. The gray dot-dashed lines indicate the energetic baselines ((n−1)A + CO). | |
{kind=link}
The right part of Fig. 3 (shown in blue) illustrates the isomerizations from μ2-O-bent structure to η2-CO2-tagged structure. The proposed pathways proceed via the LH-like mechanism, in which one CO molecule is firstly adsorbed onto one metal atom of the μ2-O-bent precursor, and then the resultant μ2-O-bent structure is transformed into a η2-CO2-tagged structure through an intrasystem CO attack on the bridging oxygen atom. The isomerization pathway involves one transition state TSn2 leading to the intermediate In1, in which a CO2 subunit is asymmetrically bound to the bridging position of the Ta–Ni axis, and second transition state TSn3 leading to the final η2-CO2-tagged structure, in which the CO2 moiety is only bound to the Taatom. Generally, the CO oxidation reaction proceeding via a LH mechanism has to satisfy two requirements: the sufficiently large carbonyl binding energy and the lower energetics of the transition states involved in the subsequent CO2 moiety formation than the initial reactants. As can be seen from Fig. 3, the attack of a chemisorbed CO ligand on the bridging O atom to form the intermediate is the bottleneck for the CO oxidation by the heteronuclear TaNiO(CO)n− complexes. This key step of the isomerization process involves the formation of one C-O bond along with the weakening of Ta-O bond. The next step, i.e., the migration of the CO2 moiety from the unsymmetrical bridging position of the Ta-Ni axis to the Ta site, involves the breaking of the Ni-O bond. That is to say, the activation of the TaONi core upon CO chemisorption is vitally important for the oxygen migration so as to facilitate the CO oxidation. However, despite the large exothermicity of CO chemisorption on the TaNiO(CO)n− (n=5–7) complexes, the formation of η2-CO2-tagged structures is impeded by the relatively high barrier of the transition states, some of which are even higher with respect to the separate reactants. This explains why the η2-CO2-tagged isomer is not observed experimentally for TaNiO(CO)n− (n=5–7).
The pathways of CO oxidation on the TaNiO(CO)7− complex are also evaluated to understand the evolution from the CO association to the CO oxidation at n=8. Fig. 4a shows the potential energy profiles of the CO oxidation reaction on the isomer 7B. Two distinct reaction channels are identified for the formation of the CO2 unit. The very first reaction step is the formation of the weakly bound complex, in which the fresh CO molecule can be either chemisorbed onto the Ta atom to form isomer 8B or physisorbed onto 7B to generate intermediate I81. As opposed to the strongly exothermic association of CO onto TaNiO(CO)n− (n=4–6) complexes, the CO adsorption on isomer 7B is almost thermoneutral, suggesting that the adsorption of CO on TaONi core presumably reaches saturation. These CO association complexes are separated by transition states TS81 and TS82, respectively, from the same intermediate I82 in which the freshly adsorbed CO is obliquely attached to the bridging oxygen atom to form a bent CO2 unit. The freshly chemisorbed CO ligand to the Ta atom bends forwards to the bridging oxygen atom, leading to the formation of C-O bond in the transition state TS81. The transition state TS82 has a structure related to the initial complex with a physisorbed CO ligand much closer to counterpart. Apparently, the pathway for CO chemisorption leading to the formation of intermediate. I82 involves an internal CO attack and belongs to the LH-like mechanism, while the other pathway for CO physisorption involves an intersystem CO attack and can be attributed to the ER-like mechanism. Note that the CO2 subunit has already been formed at this critical juncture of intermediate I82. As compared with those of TaNiO(CO)n− (n=5–7), the reaction energy barriers for the attack of CO ligand on the bridging O atom are dramatically reduced, suggesting that the TaONi core becomes significantly reactive towards CO oxidation upon additional CO chemisorption.

|
Download:
|
| Fig. 4. DFT calculated potential energy profiles for the CO oxidation reactions starting from 7B + CO (a) and 7A + CO (b). The zero-point vibration corrected energies (eV) with respect to the separated reactants are given. The dot-dashed line indicates the energetics of reactants 7A and CO, while the dashed line stands for the energetics of the isomer 7B and CO. For the sake of distinction, the C and O atoms of the freshly adsorbed CO ligand are depicted in different colors from the rest of CO ligands. | |
{kind=link}
Then the reaction proceeds further to form the more stable CO2-tagged structure through the CO2 migration. By the rotation of CO out of the TaONi core plane and shortening of Ta–Ni distance, the structure. I82 can be further isomerized to important intermediate. I83, which requires surmounting a low barrier of 0.19 eV (TS83). Thereafter, the reaction proceeds following two different channels, which both involve the migration of CO2 and the conversion of two CO ligands from the terminal to the bridging coordination. The coordination pattern evolutions of two CO ligands can happen step by step in one reaction channel, or simultaneously in the other reaction channel. The energetically more favorable pathway involves firstly an almost no barrier step (TS84) to form CO singly bridging stereochemistry, followed by a deviation of the CO2 unit away from the Ni atom and a transition from terminal to bridging coordination for another CO ligand, which is connected with the barrier of 0.14 eV (TS86). The second reaction pathway involves the breaking of the Ni-O bond and the simultaneous formation of the doubly CO bridging stereochemistry. The corresponding transition state (TS85) is 0.24 eV higher in energy than the initial reactants 7B and CO. At first glance, the oxidation reaction might be prevented due to the transition states with barriers slightly higher than the separate reactants 7B and CO. However, the heights of the involved activation barriers are much smaller with respect to the cases of TaNiO(CO)n− (n=5–7), which can be readily surmounted by thermal collision in the supersonic molecular beam [19]. In addition, the whole CO oxidation reaction is thermodynamically exothermic by 0.45 eV. Therefore, the CO oxidation on TaNiO(CO)7− via both ER- and LH-like mechanisms are facile.
For comparison, the CO oxidation reactions starting from 7A and CO are also calculated, provided that 7A exists, and the corresponding potential energy profiles are present in Fig. 4b. Firstly, the CO molecule can be physisorbed or chemisorbed to generate complex I85 or 8A, respectively. For complex 8A, the CO oxidation reaction proceeds further following the similar pathways as TaNiO(CO)n− (n=5–7), i.e., firstly the attack of a chemisorbed CO ligand on the bridging O atom to form CO2 subunit and secondly the change of coordination patterns of CO2 moiety from a bridge-type coordination to a side-on coordination on only Ta atom. While the further approaching of freshly physisorbed CO in the complex I85 to the bridging oxygen atom will give rise to the same intermediate I83, which can react following the same pathways as the abovementioned (Fig. 4a). It is demonstrated that the important intermediate I83 can be generated either from the 7A and CO via an ER-like mechanism or from 7B and CO via both an ER-like mechanism and a LH-like mechanism. Obviously, the reactions occur through an ER-like mechanism is more favorable, whereas the reactions via a LH-like mechanism may still not easily proceed, as the CO molecule will experience quite a higher reaction barrier (TS88) when the adsorbed CO approaches to the bridging oxygen atom.
As shown in Table S3 (Supporting information), the Mayer bond orders for the Ta-O bonds (1.17–1.77) in the oxygen-bridged isomers are larger than those of Ni-O bonds (0.45–0.78). The stronger the TM-O bond strength is, the higher the reaction barrier will be. The differences in the bond orders are closely correlated with the distinct energy barriers of the two transition states TSn2 and TSn3 (n=5–7) involved in the structural rearrangement from CO association to CO oxidation, suggesting that the activation of the Ta-O bond involved in the first step of the proposed pathway is critical. The natural population analysis indicates that both Ta and Ni atoms behave as electron acceptors upon the consecutive CO adsorption. The partial charges of Ni and Ta atoms reach their minimal value at n=7 and n=8, respectively, while the partial charges on the bridging O atom are almost unchanged. As a result, the charge difference between the Ni atom and the bridging O atom has a sudden change at n=7, while the abrupt decrease in the charge difference between the bridging O atom and the Ta atom occurs at n=8. The decrease of charge separation in TM-O subunits, leading to smaller ionic character of the TM-O bonds, may decrease atomic oxygen dissociation energy [19], and thus facilitates the atomic oxygen migration and the corresponding CO oxidation occurring at n=8. The variation of natural populations correlates well with the change in Mayer bond orders. The consecutive CO adsorption has minimal perturbation on Ni-O bond in terms of bond length and bond order until the TaONi core chemisorbs up to 7 CO ligands. Despite significant reduction in bond order of Ni-O bond already occurring at n=7, the oxidation reaction is still difficult, because CO multi-adsorption do not dramatically weaken the Ta-O bond until CO adsorption reaches the critical number (n=8). It is demonstrated that, in comparison with the congeneric NbNiO(CO)n− complexes at n=7 [18], one more CO ligand adsorption is needed to promote the CO oxidation on the TaNiO(CO)n− series. The current work further sheds light on the concept of CO self-promotion in the CO oxidation.
The CO adsorption energy retained by the TMO clusters is deem to be the driving force for CO oxidation [20]. The gold oxide clusters with different charge states, bearing great diversity in CO binding energy, express a preference for an ER-like or a LH-like mechanism for CO oxidation on them [20]. The LH-like mechanism is generally considered to be preferential for the CO oxidation in the gas phase [21], because the gained energy from the initial CO chemisorption in the LH-like mechanism usually is larger than that from the initial CO physisorption in ER-like mechanism. For the initial process of consecutive CO adsorption, the CO physisorption energies on TaNiO(CO)n− complexes are so small that the ER-like mechanism becomes uncompetitive relative to the LH-like mechanism. However, as shown in Fig. S3 (Supporting information), the step binding energies of CO to the O-bridged isomers monotonically decrease with the consecutive CO adsorption, and finally converge to the values close to the physisorption energies. Consequently, the chemisorption loses its competitive edge at n=8, and both ER- and LH-like mechanisms prevail for the CO oxidation reaction on TaNiO(CO)8− complex.
Herein we identified the compositional dependence of reactivity and selectivity of the reaction mechanism for the heteronuclear complexes, which is rooted in the inherent nature of transition metal components. This dramatic difference in reactivity can be attributed to the stronger TM-O bonds of the 5d metals with respect to those of the lighter 3d and 4d congeners. The dissociation energy of diatomic TaO (8.30 eV) [22] has been confirmed to be larger than the values of 6.48 and 7.53 eV for lighter VO [23] and NbO [24]. The high-level ab initio calculations with the relativistic and correlation effects taken into account have demonstrated that the fascinating chemistry of 5d metals with regard to various chemical processes such as CO oxidation and C-H bond activation is to a great extent caused by relativity [25, 26]. This same reason was invoked to elaborate on the discrepant maximum carbonyl-coordination number of the vanadium family (V+, Nb+, Ta+) [27]. The relativistic mass velocity contraction of the 6s orbital of Ta results in the contracted Ta-O bond (Table S3 in Supporting information) with respect to the corresponding Nb-O bond [18]. As a result of the 6s orbital stabilization and 5d orbital destabilization for Ta, the 4F (3d36s2) ground state of Ta is in contrast to the 6D (3d46s1) ground state of Nb. Consequently, the electron affinity of Ta atom (0.323 eV) [28] is significantly lower than those of lighter homologsV atom (0.526 eV) [28] and Nb atom (0.894 eV) [28]. Furthermore, the 6s orbital stabilization and 5d orbital destabilization for Ta are complemented by a significantly better overlap between the π orbitals of the CO ligand with the 5dπ orbitals of Ta and therefore greater πback-donation of electron density from the 5dπ orbitals of Ta, when compared to the 3dπ and 4dπ counterparts of V and Nb. Accordingly, the capability of net electron storage of Ta atom in TaNiO(CO)n− complexes is less competitive with respective to that of Nb atom in congeneric NbNiO(CO)n− [18]. As a consequence of the stronger Ta-O bond and the less competitive electron storage ability of Ta, the TaNiO(CO)n− complexes are less reactive towards CO oxidation than corresponding NbNiO(CO)n− congeners and additional CO ligand adsorption is needed to drive the CO oxidation on TaNiO(CO)n− complexes. Note that the calculated Mayer bond orders of Ta-O bond in μ2-O-bent structures of TaNiO(CO)n− (n=4–7) fall in the range of 1.32–1.52, while the those for the μ2-O-linearisomers are even larger and range from 1.60 to 1.77, suggesting the double-bond character of the Ta-O bond. As compared to the Ta+ cation forming a seven-coordinate complex [27], the maximum carbonyl-coordination number of Ta atom in the anionic TaNiO(CO)n− complexes should decrease to six. The Ni atom in TaNiO(CO)7− has reached its chemisorption saturation limit and its minimal value of partial charge. Consequently, beyond TaNiO(CO)7−, additional CO ligand can either be physisorbed in order to retain the Ta-O double bond, or be accommodated via chemisorption so that the Ta-O double bond becomes a single bond to reach the chemisorption saturation limit. In other word, both the chemisorption and physisorption on TaNiO(CO)7− are so comparable with each other that both ER- and LH-like mechanisms prevail for the CO oxidation reaction on TaNiO(CO)8− complex.
In summary, the structure evolution of the TaNiO(CO)n− (n=5–8) series is characterized by the photoelectron velocity-map spectroscopy combined with DFT calculations. Three different types of structures participate in the competition of ground state, with the μ2-O-bent structure firstly being most favor, then the μ2-O-linear structure being most preferential, and finally, the η2-CO2-tagged structure being dominant. In contrast to the CO oxidation on congeneric V-doped [15] and Nb-doped [22] nickel oxide complexes, which proceed via an LH-like mechanism as a result of the multi-adsorption of CO ligands, complementary theoretical calculations reveal that both ER- and LH-like mechanisms generally become favorable and lead to the self-promoted CO oxidation on TaNiO(CO)8−. Our findings shed new light on the role of the composition of heteronuclear metal complexes in the regulation of the reactivity and selectivity of the CO oxidation mechanism, which potentially benefits the rational design and development of the high-efficiency catalysts.
Declaration of competing interestThe authors declare no competing financial interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21603130, 21673231, 21688102 and 21873097); the Key Research Program (No. KGZD-EW-T05), the Strategic Priority Research Program (No. XDB17000000) of the Chinese Academy of Science. Zhiling Liu also gratefully acknowledges the Shanxi Province Science Foundation for Youths (No. 201901D211395), the 1331 Engineering of Shanxi Province and the Start-up Fund from Shanxi Normal University for support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:
| [1] |
A.A. Herzing, C.J. Kiely, A.F. Carley, P. Landon, G.J. Hutchings, Science 321 (2008) 1331-1335. DOI:10.1126/science.1159639 |
| [2] |
X.W. Xie, Y. Li, Z.Q. Liu, M. Haruta, W.J. Shen, Nature 458 (2009) 746-749. |
| [3] |
L. Nie, D.H. Mei, H.F. Xiong, et al., Science 358 (2017) 1419-1423. DOI:10.1126/science.aao2109 |
| [4] |
B.T. Qiao, A.Q. Wang, X.F. Yang, et al., Nat. Chem. 3 (2011) 634-641. |
| [5] |
R.J.H. Grisel, B.E. Nieuwenhuys, J. Catal. 199 (2001) 48-59. DOI:10.1006/jcat.2000.3121 |
| [6] |
N. Lopez, T.V.W. Janssens, B.S. Clausen, et al., J. Catal. 223 (2004) 232-235. DOI:10.1016/j.jcat.2004.01.001 |
| [7] |
I. Langmuir, Trans. Faraday Soc. 17 (1922) 607-620. DOI:10.1039/TF9221700607 |
| [8] |
G. Ertl, Surf. Sci. 299-300 (1994) 742-754. DOI:10.1016/0039-6028(94)90694-7 |
| [9] |
H. Schwarz, K.R. Asmis, Chem. Eur. J. 25 (2019) 2112-2126. DOI:10.1002/chem.201805836 |
| [10] |
J.B. Ma, Z.C. Wang, M. Schlangen, S.G. He, H. Schwarz, Angew. Chem. Int. Ed. 52 (2013) 1226-1230. DOI:10.1002/anie.201208559 |
| [11] |
C.X. Chi, H. Qu, L.Y. Meng, et al., Angew. Chem. Int. Ed. 56 (2017) 14096-14101. DOI:10.1002/anie.201707898 |
| [12] |
L.N. Wang, Z.Y. Li, Q.Y. Liu, et al., Angew. Chem. Int. Ed. 54 (2015) 11720-11724. DOI:10.1002/anie.201505476 |
| [13] |
X.N. Li, Z.Y. Li, H.F. Li, S.G. He, Chem. Eur. J. 22 (2016) 9024-9029. DOI:10.1002/chem.201600451 |
| [14] |
L.N. Wang, X.N. Li, L.X. Jiang, et al., Angew. Chem. Int. Ed. 57 (2018) 3349-3353. DOI:10.1002/anie.201712129 |
| [15] |
X.P. Zou, L.N. Wang, X.N. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 10989-10993. DOI:10.1002/anie.201807056 |
| [16] |
L.N. Wang, X.N. Li, S.G. He, J. Phys. Chem. Lett. 10 (2019) 1133-1138. DOI:10.1021/acs.jpclett.9b00047 |
| [17] |
X.N. Li, Z. Yuan, S.G. He, J. Am. Chem. Soc. 136 (2014) 3617-3623. DOI:10.1021/ja412608b |
| [18] |
J.M. Zhang, Y. Li, Z.L. Liu, et al., J. Phys. Chem. Lett. 10 (2019) 1566-1573. DOI:10.1021/acs.jpclett.9b00205 |
| [19] |
M.L. Kimble, N.A. Moore, G.E. Johnson, et al., J. Chem. Phys. 125 (2006) 204311. DOI:10.1063/1.2371002 |
| [20] |
C. Bürgel, N.M. Reilly, G.E. Johnson, et al., J. Am. Chem. Soc. 130 (2008) 1694-1698. DOI:10.1021/ja0768542 |
| [21] |
R.J. Baxter, P. Hu, J. Chem. Phys. 116 (2002) 4379-4381. DOI:10.1063/1.1458938 |
| [22] |
S. Smoes, J. Drowart, C.E. Myers, J. Chem. Thermodyn. 8 (1976) 225-239. DOI:10.1016/0021-9614(76)90100-2 |
| [23] |
G. Balducci, G. Gigli, M. Guido, J. Chem. Phys. 79 (1983) 5616-5622. DOI:10.1063/1.445682 |
| [24] |
H.-P. Loock, B. Simard, S. Wallin, C. Linton, J. Chem. Phys. 109 (1998) 8980-8992. DOI:10.1063/1.477568 |
| [25] |
M. Dolg, H. Stoll, H. Preuss, R.M. Pitzer, J. Phys. Chem. 97 (1993) 5852-5859. DOI:10.1021/j100124a012 |
| [26] |
H. Schwarz, Angew. Chem. Int. Ed. 42 (2003) 4442-4454. DOI:10.1002/anie.200300572 |
| [27] |
A.M. Ricks, Z.D. Reed, M.A. Duncan, J. Am. Chem. Soc. 131 (2009) 9176-9177. DOI:10.1021/ja903983u |
| [28] |
C.S. Feigerle, R.R. Corderman, S.V. Bobashev, W.C. Lineberger, J. Chem. Phys. 74 (1981) 1580-1598. DOI:10.1063/1.441289 |