 2021, Vol. 32
2021, Vol. 32 


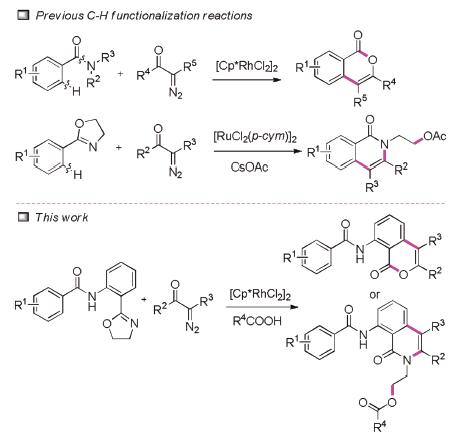
Rhodium-catalysed C-H functionalization has emerged as an effective tool in organic synthesis for the construction of diversified heterocyclic systems [1]. C-H bond functionalization [2] is an attractive alternative to classical cross-coupling reactions (which usually require organohalides and organometallic reagents) due to the abundance and relatively low cost of various hydrocarbons. Thus, merging C-H functionalizations with other efficient and sustainable approaches like multicomponent reactions or cascade transformations, is expected to be an ideal tool in organic synthesis for rapid construction of complex molecules from simple starting materials [3].
Recently, diazo compounds as good partners for C-H activation couplings or cyclizations, are widely used in synthetic organic chemistry (Scheme 1) [4]. In 2012, Yu reported the Rh(III)-catalysed intermolecular cross-coupling of diazomalonates with arene C-H bonds [5]. Subsequent work demonstrated the Rh(III)-catalysed intermolecular cross-coupling of diazo compounds [6]. Although the aforementioned methods are encouraging, a straightforward C-H activation strategy to access isocoumarin and isoquinolinone scaffolds with different substitutions remains highly desireable [7]. To the best of our knowledge, one-pot ring-opening/ring-closure of oxazole via Rh(III)-catalyzed C-H activation/cyclization of aromatics with diazo compounds has not been reported.

|
Download:
|
| Scheme 1. Rh(III)/Ru(II)-catalysed C–H activation/cyclization using diazo compounds as cyclization partners. | |
{kind=link}
Isocoumarin and isoquinolinone are heterocyclic compounds [8] that provide the key structural motif for a variety of biologically and pharmacologically important natural products. Despite the vast array of methods to synthesize isocoumarins and isoquinolinones, new methods that efficiently access highly decorated substituted isocoumarins and isoquinolinones remain desirable [9]. Herein, we disclose an acid-controlled [10], one-pot, ring-opening/ring-closure pathway [11] for combining phenyloxazoles with diazos to rapidly and economically stepwise construct polycyclic isocoumarins in high yields.
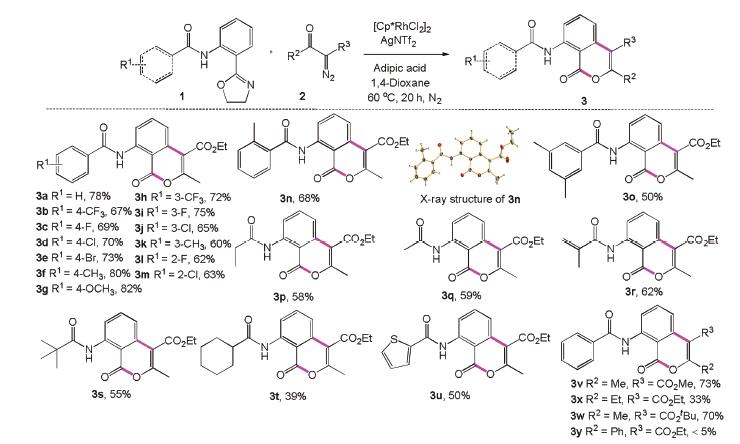
The envisaged functionalization/cyclization sequence was investigated and optimized using N-(2-(4,5-dihydrooxazol-2-yl)phenyl)benzamide (1a) and ethyl 2-diazo-3-oxobutanoate (2a) as the prototype substrate combination (see Supporting information for details). To our delight, excellent conversions of 1a were achieved when the reaction was carried out at 100℃ for 20h in the presence of [Cp*RhCl2]2 (2.5mol%), AgSbF6 (10mol%) and PivOH (2.0 equiv.) under air and using 1,4-dioxane as the solvent. The corresponding cyclization products 3a and 4a, were isolated in 32% and 40% yields, respectively (see Supporting information for details). To improve the yield and selectivity of 3a, we changed the acid amount. Disappointingly, we found that reducing the amount of acid had little effect on either yield or selectivity. However, there is no doubt that acid plays a decisive role in this process. Several acids were subsequently investigated and to our satisfaction, good selectivities and yields for 3a were obtained when a binary acid was involved in the reaction system. Propanedioic acid and sebacic acid were both inferior to adipic acid (see Supporting information for further details) [12]. We tried different additives to improve the conversion and AgNTf2 was more suitable than others for this transformation. Those results demonstrated that the reaction could be conducted at 60℃ under an inert atmosphere with yields of 3a reaching 78% yield. Compared with another commonly used organic phase solvent DCE, 1,4-dioxane gave a better yield (see Supporting information for further details). Once an efficient synthesis of 3a had been established, our attention turned to the preferential formation of 4a. Solvents effects were initially examined and when the reaction was carried out in DMSO and toluene, only trace amounts of the desired products were obtained. However, when THF was used as the solvent, 4a was obtained in 55% yield. Lastly, the temperature was changed to 80℃, which resulted in excellent yields of 4a (see Supporting information for further details). Further studies on the catalysts revealed that [Cp*RhCl2]2 was the most effective, with no isocoumarins products detected using Rh2(OAc)4, [Rh(cod)Cl]2, [Ru(p-cym)Cl2]2 and [Cp*IrCl2]2 as catalysts (see Supporting information for further details).
As shown in Scheme 2, the scope and limitations for the formation of isocoumarins 3a–3z were investigated using systematic variation of phenyloxazoles (1a–1u) and diazo compounds (2a–2d). Ethyl 2-diazo-3-oxobutanoate (2a) was chosen as a representative coupling partner. Phenyloxazoles bearing a variety of substituted groups on the benzene ring, which included both electron-rich groups (methyl, methoxy) and electron-deficient groups (trifluoromethyl, fluoride, chloride and bromide), exhibited high reactivity with 2a under the optimised reaction conditions (3a-3n, 60%–82%). Moreover, the chemical structure of 3n was unambiguously confirmed by X-ray crystallography (Crystal Structure submitted to Cambridge Cystallographic Data Centre. CCDC: 1961003). Furthermore, substrate bearing 3,5-dimethyl substituent (1o) was also well tolerated, furnishing the required isocoumarin in moderate yield (3o, 50%). Additionally, the less sterically demanding wide range of alkyl substituted such as ethyl and methyl, also provided the corresponding product in good yield (3p, 58% and 3q, 59%). Moreover, a wide range of alkyl substituted phenyloxazoles performed well and furnished the corresponding products in satisfactory yields (3r, 62%, 3s, 55% and 3t, 39%). Heterocyclic derivatives were also well tolerated in this transformation, and moderate yields of the corresponding products 3u were obtained (3u, 50%).

|
Download:
|
| Scheme 2. Substrate scope. Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), [Cp*RhCl2]2 (2.5 mol%), AgNTf2 (10 mol%), 1,4-dioxane (2 mL), 20 h, under N2 atmosphere. Isolated yields. | |
{kind=link}
Next, we investigated the scope and reactivity of diazo compounds using N-(2-(4,5-dihydrooxazol-2-yl)phenyl)-benzamide (1a) as a representative coupling partner. When R3 was a methyl ester or a tertiary-butyl ester, the reaction proceeded smoothly and gave the corresponding products in 73% and 70% yields, respectively (3v and 3w). However, the ethyl substituted substrate (2c) gave the desired product 3x was obtained in only 33% yield. Furthermore, the reaction did not produce the corresponding product when the phenyl substituted substrate (2d) was used, probably due to the steric hindrance.
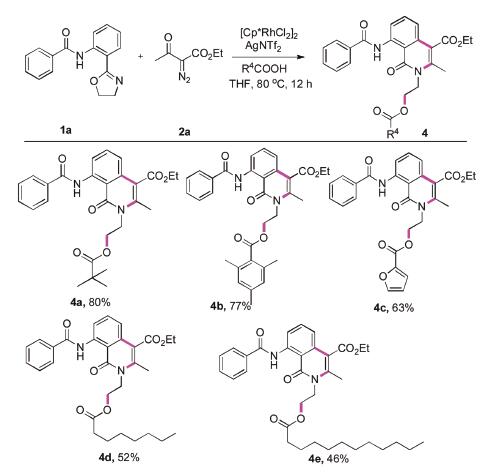
Finally, we explored the scope of this Rhodium-catalysed cyclization of N-(2-(4,5-dihydrooxazol-2-yl)phenyl)benzamide (1a) and ethyl 2-diazo-3-oxobutanoate (2a) with a variety of commercially available acids to afford the corresponding isoquinolinones. To our delight, we observed that both alkyl, aryl and heterocyclic acids were well-tolerated (Scheme 3, 4a-4c). In particular, when long-chain-fatty-acids were participated, the reaction still took place (4d and 4e). Synthesis of these compounds certainly have applications in the medical field. In fact, isoquinolinones were formed the core of several medicines currently in development.

|
Download:
|
| Scheme 3. Substrate scope. Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), [Cp*RhCl2]2 (2.5 mol%), AgNTf2 (10 mol%), THF (2 mL), 12 h. Isolated yields. | |
{kind=link}
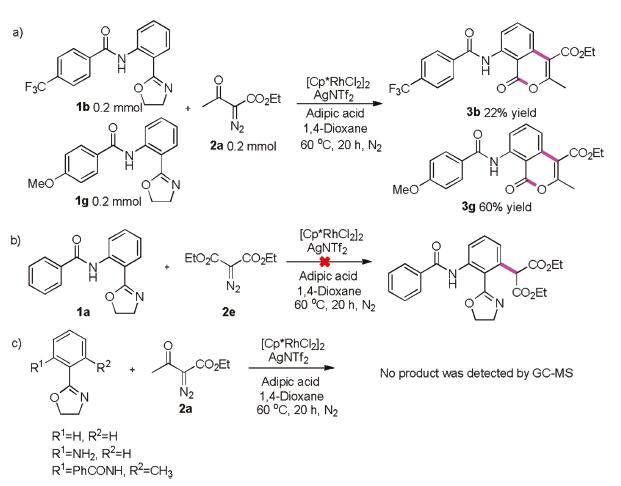
To better understand the versatile reactivity of Rh(III)-catalysed C–H activation and to gain insight into the mechanism of this transformation, we conducted several control experiments. The competition experiment between 1b and 1g was performed under standard conditions and gave products 3b and 3g in 22% and 60% yields, respectively (Scheme 4a). These results indicated that electron-donating substituents were favoured for this transformation. Furthermore, when we used diethyl 2-diazomalonate 2e, the reaction did not proceed (Scheme 4b). This implied that the ketone plays an important role in this lactonization progress. To investigate the role of amides, we replaced amides with hydrogen and amidogen groups, however, no target products were obtained. This led us to conclude that the amide group was necessary (Scheme 4c). Similarly, Rh(III)-catalysed ortho-positions C-H activation was proved (Scheme 2c).

|
Download:
|
| Scheme 4. Mechanistic studies. | |
{kind=link}
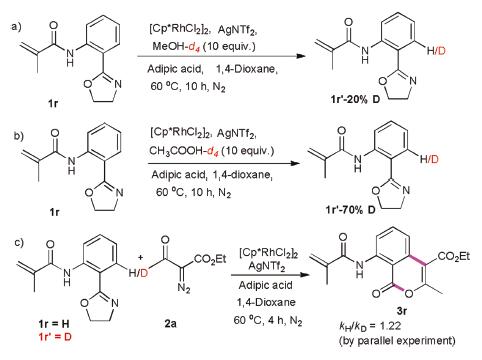
To further probe the C–H activation process, we carried out kinetic isotope effect (KIE) experiments. To eliminate the interference of other hydrogen of benzene ring, we choose 1r as the model substrate for KIE experiments. Methanol-d4 and acetic acid-d4 were separately subjected to the optimized reaction conditions over 10h. We obtained the products 1r'-20%D and 1r'-70%D at the ortho-positions (Schemes 5a and b). These results showed that acid is beneficial for C–H metalation. A low kH/kD (1.22) value was obtained for parallel experiments, and indicated that C–H cleavage was not the rate-determining step (Scheme 5c).

|
Download:
|
| Scheme 5. Kinetic isotope effect experiments. | |
{kind=link}
Based on these preliminary experimental results and literature precedents [4, 13-15], we propose a plausible mechanism in Scheme 6. First, the coordination of 1a with Rh(III) forms Rh(III) complexes A, which undergo a reversible rollover cyclometalation to give complexes B. Subsequent combination with ethyl 2-diazo-3-oxobutanoate 2a produces a rhodium-carbene intermediate C. Migratory insertion of the Rh-C bond into the activated carbene forms the six-membered rhodacycle D. The intermediate E produces 3a via enolization and esterification with the release of ethyleneimine (Scheme 6, path a). Meanwhile, E produces 4a by nucleophilic attack through the intermediate H in the presence of PivOH (Scheme 6, path b).

|
Download:
|
| Scheme 6. Plausible mechanism. | |
{kind=link}
In conclusion, we have developed a one-pot, ring-opening/ring-closure, Rh(III)-catalysed, C–H functionalization of phenyloxazoles with diazo compounds to produce isocoumarins and isoquinolinones. This transformation features acid-controlled chemodivergent annulation, operational simplicity and excellent functional group tolerance. Further applications of this Rh(III)-catalytic system to other more challenging C-H functionalizations continue in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.06.011.
| [1] |
(a) X. Guo, J. Han, Y. Liu, et al., J. Org. Chem. 82 (2017) 11505-11511; (b) N. Lv, Z. Chen, Y. Liu, Z. Liu, Y. Zhang, Adv. Synth. Catal. 361 (2019) 4140-4146; (c) S. Zhang, B. Wang, X. Jia, Y. Yuan, Adv. Synth. Catal. 361 (2018) 451-455; (d) Q. Wang, Y. Xu, X. Yang, Y. Li, X. Li, Chem. Commun. 53 (2017) 9640-9643; (e) L. Kong, X. Han, X. Li, Chem. Commun. 55 (2019) 7339-7342. |
| [2] |
(a) Z. Huang, H.N. Lim, F. Mo, M.C. Young, G. Dong, Chem. Soc. Rev. 44 (2015) 7764-7786; (b) Y. Huang, W.J. Pan, Z.X. Wang, Org. Chem. Front. 6 (2019) 2284-2290; (c) L. Zhang, J. Chen, J. Chen, et al., Tetrahedron Lett. 60 (2019) 1053-1056. |
| [3] |
(a) P.B. Arockiam, C. Bruneau, P.H. Dixneuf, Chem. Rev. 112 (2012) 5879-5918; (b) Y. Qin, L. Zhu, S. Luo, Chem. Rev. 117 (2017) 9433-9520; (c) Y. Xu, L. Zhang, M. Liu, et al., Org. Biomol. Chem. 17 (2019) 8706-8710; (d) Y. Li, F. Wang, S. Yu, X. Li, Adv. Synth. Catal. 358 (2016) 880-886; (e) J.A. Ellman, L. Ackermann, B.F. Shi, J. Org. Chem. 84 (2019) 12701-12704; (f) D.Y. Wang, S.H. Guo, G.F. Pan, et al., Org. Lett. 20 (2018) 1794-1797; (g) M.N. Zhao, Z.H. Ren, D.S. Yang, Z.H. Guan, Org. Lett. 20 (2018) 1287-1290. |
| [4] |
(a) X.G. Li, M. Sun, K. Liu, Q. Jin, P.N. Liu, Chem. Commun. 51 (2015) 2380-2383; (b) G.S. Kumar, N.P. Khot, M. Kapur, Adv. Synth. Catal. 361 (2019) 73-78; (c) L. Kong, X. Han, X. Li, Chem. Commun. 55 (2019) 7339-7342; (d) M. Wang, L. Kong, Q. Wu, X. Li, Org. Lett. 20 (2018) 4597-4600; (e) X. Chen, G. Zheng, G. Song, X. Li, Adv. Synth. Catal. 360 (2018) 2836-2842; (f) B. Shen, B. Wan, X. Li, Angew. Chem. 130 (2018) 15760-15764; (g) S.Y. Yan, P.X. Ling, B.F. Shi, Adv. Synth. Catal. 359 (2017) 2912-2917. |
| [5] |
W.W. Chan, S.F. Lo, Z. Zhou, W.Y. Yu, J. Am. Chem. Soc. 134 (2012) 13565. |
| [6] |
(a) Z. Shi, D.C. Koester, M. Boultadakis-Arapinis, F. Glorius, J. Am. Chem. Soc. 135 (2013) 12204-12207; (b) S.S. Cui, Y. Zhang, D. Wang, Q. Wu, Chem. Sci. (2013) 3912-3916; (c) Z. Shi, D.C. Koester, M. Boultadakis-Arapinis, F. Glorius, J. Am. Chem. Soc. 135 (2013) 12204-12207; (d) X. Yu, S. Yu, J. Xiao, B. Wan, X. Li, J. Org. Chem. 78 (2013) 5444-5452. |
| [7] |
(a) Y.H. Wang, G. Qiu, H. Zhou, W. Xie, J.B. Liu, Tetrahedron 75 (2019) 3850-3855; (b) V. Pirovano, M. Marchetti, J. Carbonaro, et al., Dye. Pigment. 173 (2020) 107917-107930; (c) X. Chen, Y. Liu, RSC Adv. 7 (2017) 37839-37843. |
| [8] |
(a) G.D. Xu, Z.Z. Huang, Org. Lett. 19 (2017) 6265-6267; (b) M. Zheng, L. Huang, Q. Tong, W. Wu, H. Jiang, Eur. J. Org. Chem. 2016 (2016) 663-667; (c) N.A. Danilkina, L.Y. Gurskaya, A.V. Vasilyev, I.A. Balova, Eur. J. Org. Chem. 2016 (2016) 739-747. |
| [9] |
(a) F. Lin, Y. Shen, Y. Zhang, et al., Chem. Eur. J. 24 (2018) 551-555; (b) T. Izumi, Y. Nishimoto, K. Kohei, A. Kasahara, J. Heterocycl. Chem. 27 (1990) 1419-1424; (c) S.L. Zhang, Z.L. Yu, Org. Biomol. Chem. 14 (2016) 10511-10515. |
| [10] |
(a) C. Hu, G. Hong, Y. He, et al., J. Org. Chem. 83 (2018) 4739-4753; (b) S. Koley, T. Chanda, B.J. Ramulu, S. Chowdhury, M.S. Singh, Adv. Synth. Catal. 358 (2016) 1195-1201. |
| [11] |
(a) J.W. Ren, Q.L. Zhao, J.A. Xiao, et al., Chem. Eur. J. 25 (2019) 4673-4677; (b) X. Guo, Q. Xing, K. Lei, et al., Adv. Synth. Catal. 359 (2017) 4393-4398; (c) L.A. Perego, S. Wagschal, R. Grüber, et al., Adv. Synth. Catal. 361 (2019) 151-159. |
| [12] |
(a) S. Capelli, D. Motta, C. Evangelisti, et al., ChemCatChem11 (2019)3075-3084; (b) J.W. Jiang, C.C. Tu, C.H. Chen, Y.C. Lin, ChemCatChem 10 (2018) 5449-5458. |
| [13] |
(a) Z. Yang, L. Jie, Z. Yao, Z. Yang, X. Cui, Adv. Synth. Catal. 361 (2019) 214-218; (b) Z. Yang, X. Lin, L. Wang, X. Cui, Org. Chem. Front. 4 (2017) 2179-2183; (c) R. Zhu, G. Cheng, C. Jia, L. Xue, X. Cui, J. Org. Chem. 81 (2016) 7539-7544; (d) Y. Yu, C. Kuai, R. Chauvin, et al., J. Org. Chem. 82 (2017) 8611-8616; (e) J. Yu, S. Wen, D. Ba, et al., Org. Lett. 21 (2019) 6366-6369; (f) T. Jeong, S.H. Lee, N.K. Mishra, et al., Adv. Synth. Catal. 359 (2017) 2329-2336. |
| [14] |
(a) G.J. Li, Y.L. Pan, Y.L. Liu, H.F. Xu, J.Z. Chen, Org. Lett. 21 (2019) 1740-1743; (b) K. Qiao, X. Yuan, L. Wan, et al., Green Chem. 19 (2017) 5789-5793; (c) C.C. Yuan, X.L. Chen, J.Y. Zhang, Y.S. Zhao, Org. Chem. Front. 4 (2017) 1867-1871; (d) L. Wan, K. Qiao, X. Yuan, et al., Adv. Synth. Catal. 359 (2017) 2596-2604. |
| [15] |
(a) Y. Liu, J. Wu, B. Qian, Y. Shang, Org. Biomol. Chem. 17 (2019) 8768-8777; (b) M. Gao, Y. Yang, H. Chen, B. Zhou, Adv. Synth. Catal. 360 (2018) 100-105; (c) S. Dhole, W.J. Chiu, C.M. Sun, Adv. Synth. Catal. 361 (2019) 2916-2925; (d) Y.N. Aher, D.M. Lade, A.B. Pawar, Chem. Commun. 54 (2018) 6288-6291; (e) M.N. Rashed, S. Siddiki, A.S. Touchy, et al., Chem. Eur. J. 25 (2019) 10594-10605; (f) X. Chen, S. Hu, R. Chen, et al., RSC Adv. 8 (2018) 4571-4576. |