 2021, Vol. 32
2021, Vol. 32 


Since the discovery of discorhabdin C (1, Fig. 1) by Blunt and Muro and coworkers in 1986, more than 100 pyrroloiminoquinone (PIQ)-related alkaloids have been identified, and most of them were isolated from marine sponges, and a few were collected from terrestrial organisms [1]. The majority of these alkaloids displayed prominent biological activities, including antitumor [2], antimicrobial [3], antiviral [3a], antimalarial [3a, 4], immunomodulatory [5], and anti-feeding activities [6]. Besides the characteristic core PIQ ring, this group of natural products usually possess additional fused, spiro, or bridged carbocycles or heterocycles (1-5, Fig. 1) [1b, 7]. The challenging structures and remarkable biological activities of these alkaloids have garnered intense interests from synthetic chemists [8]. Although many methods for the synthesis of PIQ units have been developed [9], the strategies for the construction of other attached ring systems are rather underdeveloped.

|
Download:
|
| Fig. 1. Selected PIQ alkaloids. | |
{kind=link}
Atkamine (6, Fig. 1) is a brominated PIQ alkaloid isolated from the cold, deep water Alaskan sponge Latrunculia sp. by Zou and Hamann in 2013 [10]. Structurally, atkamine features an unprecedented heptacyclic scaffold containing a PIQ, a dihydrobenzothiophene, a bridged azepane ((8-oxa-2-azabicyclo[3.2.1]oct-3-ene), a long (Z)-olefin sidechain, and four contiguous stereocenters. Of particular note is a diverse array of heteroatoms within the molecule, including nitrogen, sulfur, oxygen, and bromine. The absolute configuration of atkamine was determined by electronic circular dichroism (ECD) experiments and density functional theory (DFT) calculation. Furthermore, Hamman et al. proposed a biosynthetic pathway of atkamine in which two amino acids (tryptophan and tyrosine) and (Z)-15-docosenoic acid are involved, though several key steps have not yet been clarified. The bioactivity of atkamine was not reported, probably due to the low natural abundance (0.000125% isolation yield). The unique polycyclic structure and unknown bioactivity make atkamine an attractive and challenging target for synthetic chemists. To date, there was only one synthetic study toward 6 despite its intriguing structure [11]. As part of our continuing interest in developing novel synthetic strategies toward the halogenated marine natural products [12], we herein report our synthetic progress toward this complex brominated PIQ alkaloid.
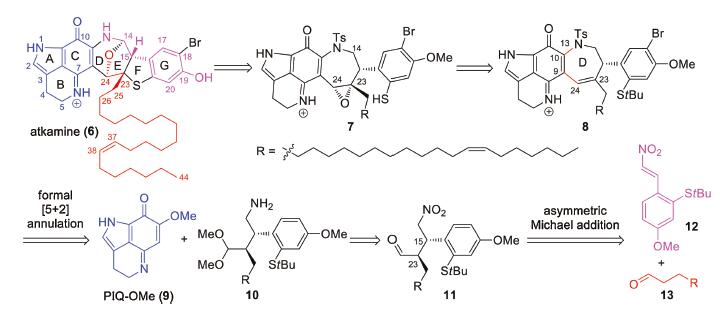
The unprecedented structure and preliminary biosynthetic proposal of atkamine inspired us to pursue a modular synthetic strategy. As depicted in Scheme 1, we envisioned that atkamine (6) could arise from epoxide precursor 7 through intramolecular epoxide opening by benzenethiol to generate the dihydrothiophene ring (F ring) and subsequent oxidative tetrahydrofuran (D ring) formation. Then 7 could be obtained from olefin 8 by epoxidation. The azepene D ring in 8 could be forged by a formal [5 + 2] annulation of methoxy pyrroloiminoquinone (PIQ-OMe) (9) and amino acetal 10 (forming C9-C24 and C10-N13 bonds, highlighted with red lines). Several synthetic approaches to 9 have been reported [9]. 10 could be obtained from 11 by acetalization of the aldehyde group and reduction of the nitro group. The retrosynthetic scission of the C15-C23 bond of 11 led to β-nitrostyrene 12 and aldehyde 13, which could be connected by an asymmetric Michael addition reaction [13]. As a whole, similar to Hamman's biosynthetic proposal, we anticipated atkamine could be assembled with three fragments, PIQ-OMe 9, nitrostyrene 12, and aldehyde 13 in a convergent manner.

|
Download:
|
| Scheme 1. Retrosynthetic analysis of atkamine (6). | |
{kind=link}
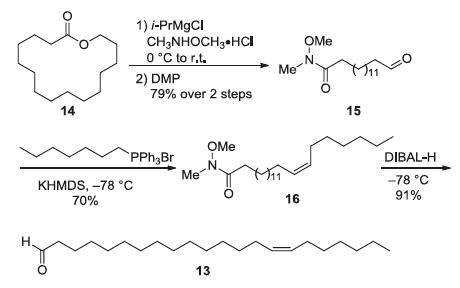
Our synthesis commenced with the synthesis of the long-chain aliphatic enaldehyde 13 from macrolide 14 (Scheme 2). The lactone ring was opened to form a Weinreb amide intermediate, which was further oxidized to generate aldehyde 15 in 79% overall yield. Then the Z-olefine 16 was smoothly constructed via Wittig reaction, which underwent reduction with DIBAL-H to afford the desired aldehyde 13 in high yield.

|
Download:
|
| Scheme 2. Synthesis of aldehyde 13. | |
{kind=link}
Then we focused on the synthesis of the amino acetal fragment. To supply a large amount of substrate for a thorough exploration of the formal [5 + 2] annulation reaction, a simplified amino acetal 21 was used (Scheme 3). To begin, β-nitrostyrene 18 was prepared from p-anisaldehyde 17 and nitromethane through Henry condensation [14]. After a brief survey of reaction conditions, we were delighted to find that when β-nitrostyrene 18 and aldehyde 13 were treated with l-proline lithium salt in tBuOMe at −20 ℃, the nitroaldehyde 19 was obtained in 87% yield and 93% ee [13c]. Then 19 was converted to the corresponding acetal 20 by treatment with pTSA and trimethylorthoformate. Finally, the nitro group in 20 was smoothly reduced to an amino group by LiAlH4 to deliver the desired amino acetal 21 in 62% yield. Interestingly, when 20 was treated with NaBH4 in the presence of NiCl2, the double bond in the aliphatic chain was saturated while the nitro group was also reduced to generate an HCl salt (22) of the corresponding amino acetal [15]. Fortunately, we got a single crystal structure of 22 (CCDC No. 2001009), which confirmed the absolute configuration of the C15 and C23 stereocenters.

|
Download:
|
| Scheme 3. Synthesis of aminoacetal 21. | |
{kind=link}
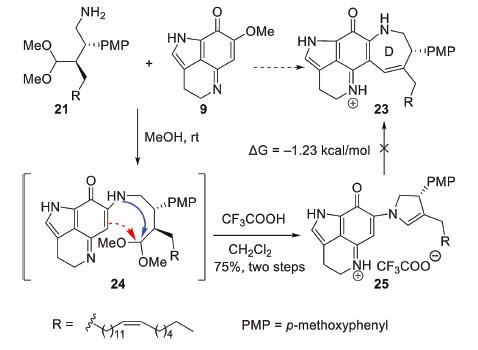
With the amino acetal 21 and PIQ-OMe 9 in hand, our investigation was then focused on the key formal [5 + 2] annulation (Scheme 4). We anticipated that the amino group of 21 would first undergo a Michael addition-elimination cascade with PIQ-OMe 9 to generate the coupling intermediate 24, which could undergo an intramolecular aldol condensation between the enamine group and the acetal group to form the azepene D ring. To our delight, the coupling reaction of 21 and 9 proceeded smoothly in MeOH at room temperature; however, further treatment of the crude coupling product with TFA only gave dihydropyrrol compound 25 as the sole product. Although DFT calculations showed that the conversion of 25 to the desired azepene product 23 was a thermodynamically viable process (ΔG = -1.23 kcal/mol) (Supporting information for details), we tried many Bronsted acids or Lewis acids at room temperature or upon heating and none of them worked [16]. To evaluate the influence of the steric hindrance of the substrate, we also synthesized the amino acetals devoid of either PMP group or long aliphatic side chain (Supporting information for details). Unfortunately, in these cases, only a tiny amount of the desired azepene products were observed under the above conditions.

|
Download:
|
| Scheme 4. Attempts for the formal [5 + 2] annulation. DFT calculation was carried out on a model substrate (R = Et) for simplicity. | |
{kind=link}
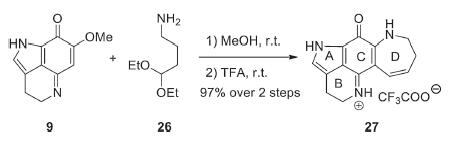
To further examine the feasibility of such a challenging formal [5 + 2] annulation strategy, we investigated this transformation with PIQ-OMe 9 and 4,4-diethoxybutanamine 26, an amino acetal without any extra substituent (Scheme 5). To our great delight, when the above conditions were employed, the azepene ring was successfully constructed to deliver tetracyclic compound 27 in 97% overall yield.

|
Download:
|
| Scheme 5. A successful model for the formal [5 + 2] annulation. | |
{kind=link}
In summary, we have developed a modular strategy towards the complex marine pyrroloiminoquinone alkaloid atkamine, featuring a formal [5 + 2] annulation for the construction of the central azepene D ring and an asymmetric Michael addition to couple the long-chain aldehyde fragment and β-nitrostyrene fragment. The efficient synthesis of the long-chain aliphatic aldehyde 13 and chiral amino acetal 21 have been achieved. A simplified tetracyclic intermediate 27 bearing the core structure of atkamine has been constructed through the formal [5 + 2] annulation. More exploration of the key [5 + 2] annulation in the complex substrate system and the further elaboration of the advance intermediate 27 to atkamine are currently undergoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge the National Natural Science Foundation of China for financial support of this work by grants (Nos. 21772164 and 21572187). We thank the National Found for Fostering Talents of Basic Science (NFFTBS, No. J1310024), New Century Excellent Talents in University of Fujian Province (NECTFJ) and Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) for support.
Appendix A.Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.06.008.
| [1] |
(a) N.B. Perry, J.W. Blunt, J.D. McCombs, M.H.G. Munro, J. Org. Chem. 51 (1986) 5476-5478; (b) J.F. Hu, H. Fan, J. Xiong, S.B. Wu, Chem. Rev. 111 (2011) 5465-5491; (c) E.M. Antunes, B.R. Copp, M.T. Davies-Coleman, T. Samaai, Nat. Prod. Rep. 22 (2005) 62-72. |
| [2] |
(a) L. Legentil, L. Benel, V. Bertrand, B. Lesur, E. Delfourne, J. Med. Chem. 49 (2006) 2979-2988; (b) E. Delfourne, Anticancer Agents Med. Chem. 8 (2008) 910-916; (c) A. Rives, B. Le Calvé, T. Delaine, et al., Eur. J. Med. Chem. 45 (2010) 343-351. |
| [3] |
(a) M.K. Na, Y. Ding, B. Wang, et al., J. Nat. Prod. 73 (2010) 383-387; (b) E. Dolušić, P. Larrieu, C. Meinguet, et al., Bioorg. Med. Chem. Lett. 23 (2013) 47-54; (c) B. Nijampatnam, S. Dutta, S.E. Velu, Chin. J. Nat. Med. 13 (2015) 561-577. |
| [4] |
R.A. Davis, M.S. Buchanan, S. Duffy, et al., J. Med. Chem. 55 (2012) 5851-5858. DOI:10.1021/jm3002795 |
| [5] |
S.P. Gunasekera, P.J. McCarthy, R.E. Longley, et al., J. Nat. Prod. 62 (1999) 173-175. DOI:10.1021/np980293y |
| [6] |
A. Yang, B.J. Baker, J. Grimwade, A. Leonard, J.B. McClintock, J. Nat. Prod. 58 (1995) 1596-1599. DOI:10.1021/np50124a020 |
| [7] |
(a) N.B. Perry, J.W. Blunt, M.H.G. Munro, Tetrahedron 44 (1988) 1727-1734; (b) J. Kobayashi, J.F. Cheng, M. Ishibashi, et al., Tetrahedron Lett. 28 (1987) 4939-4942; (c) Y. Zou, X. Wang, J. Sims, et al., J. Am. Chem. Soc. 141 (2019) 4338-4344; (d) D.C. Radisky, E.S. Radisky, L.R. Barrows, et al., J. Am. Chem. Soc. 115 (1993) 1632-1638; (e) G.J. Hooper, M.T. Davies-Coleman, M. Kelly-Borges, P.S. Coetzee, Tetrahedron Lett. 37 (1996) 7135-7138. |
| [8] |
(a) Y. Kita, H. Tohma, M. Inagaki, K. Hatanaka, T. Yakura, J. Am. Chem. Soc. 114 (1992) 2175-2180; (b) H. Tohma, Y. Harayama, M. Hashizume, et al., J. Am. Chem. Soc. 125 (2003) 11235-11240; (c) M. Iwao, O. Motoi, T. Fukuda, F. Ishibashi, Tetrahedron 54 (1998) 8999-9010; (d) A. Rives, T. Delaine, L. Legentil, E. Delfourne, Tetrahedron Lett. 50 (2009) 1128-1130. |
| [9] |
(a) J.D. White, K.M. Yager, T. Yakura, J. Am. Chem. Soc. 116 (1994) 1831-1838; (b) E.V. Sadanandan, S.K. Pillai, M.V. Lakshmikantham, et al., J. Org. Chem. 60 (1995) 1800-1805; (c) D. Roberts, J.A. Joule, M.A. Bros, M. Alvarez, J. Org. Chem. 62 (1997) 568-577; (d) Y. Kita, M. Egi, T. Takada, H. Tohma, Synthesis 5 (1999) 885-897; (e) K. Inoue, Y. Ishikawa, S. Nishiyama, Org. Lett. 12 (2010) 436-439; (f) M.W. Smith, I.D. Falk, H. Ikemoto, N.Z. Burns, Tetrahedron 75 (2019) 3366-3370. |
| [10] |
Y. Zou, M.T. Hamann, Org. Lett. 15 (2013) 1516-1519. DOI:10.1021/ol400294v |
| [11] |
H. Li, Ph. D. Thesis, Shanghai Institute of Organic Chemistry, Shanghai, 2014.
|
| [12] |
(a) C. Liu, R. Chen, Y. Shen, et al., Angew. Chem. Int. Ed. 56 (2017) 8187-8190; (b) R. Chen, L. Li, N. Lin, et al., Org. Lett. 20 (2018) 1477-1480. |
| [13] |
(a) L. Zu, H. Xie, H. Li, J. Wang, W. Wang, Adv. Synth. Catal. 349 (2007) 2660-2664; (b) Y. Hayashi, T. Itoh, M. Ohkubo, H. Ishikawa, Angew. Chem. Int. Ed. 47 (2008) 4722-4724; (c) K. Xu, S. Zhang, Y. Hu, Z. Zha, Z. Wang, Chem. Eur. J. 19 (2013) 3573-3578; (d) T. Schnitzer, A. Budinská, H. Wennemers, Nat. Catal. 3 (2020) 143-147; (e) H. Gotoh, H. Ishikawa, Y. Hayashi, Org. Lett. 9 (2007) 5307-5309; (f) Y. Chi, L. Guo, N.A. Kopf, S.H. Gellman, J. Am. Chem. Soc. 130 (2008) 5608-5609. |
| [14] |
J.M. Lopchuk, R.P. Hughes, G.W. Gribble, Org. Lett. 15 (2013) 5218-5221. DOI:10.1021/ol402385v |
| [15] |
S. Yakabe, M. Hirano, T. Morimoto, Tetrahedron Lett. 41 (2000) 6795-6798. DOI:10.1016/S0040-4039(00)01084-4 |
| [16] |
(a) T. Kotipalli, D. Janreddy, V. Kavala, et al., RSC Adv. 4 (2014) 47833-47840; (b) G.P. Tokmakov, I.I. Grandberg, Tetrahedron 51 (1995) 2091-2098; (c) Z. Zhang, D. Wang, B. Wang, et al., Tetrahedron 69 (2013) 9063-9067. |