 2021, Vol. 32
2021, Vol. 32 


Self-assembly of ordered porous networks have been proven to be well suited for the functionalization of surfaces and, thus, has become a topic of elaborate research [1-4]. Traditionally, carboxyl acid and its derivatives are used to build H-bonded molecular networks since the carboxyl groups can serve as both hydrogen bond donors and acceptors. One of the most extensively studied models is trimesic acid (TMA), a planar molecule with three-fold symmetry consisting of three carboxyl groups attached to a central benzene ring. TMA is known to assemble in various adsorbate structures, and the most characteristic motif identified therein is chicken-wire and flower-like network structures formed through the dimerization and the trimerization of carboxyl groups at the solid-liquid interface [5-11]. The monolayer morphology of carboxyl acid is primarily governed by the structure and functional groups of the molecules, and is also influenced by the type of solvent, substrate, light irradiation, temperature, and concentration [12-17]. The influences of these factors have been studied comprehensively in earlier works [18-20]. However, there are much less work on the effects of the concentration of carboxyl acid on the self-assembly structure.
Controllable structural transformations of molecular nanoarchitectures in response to external stimuli have received considerable attention, as they are promising means to modulate the functionality of surfaces and interfaces [21-24]. Scanning tunneling microscopy (STM) uses a strong electric field between a sharp metallic tip and a flat conductive surface and provides a seamless experimental setup to manipulate the diffusion and arrangement of atoms and molecules at nanoscale [25-27]. Due to the atomically sharp nature of the STM tip and a few-angstrom-wide gap between the tip and the substrate, a highly localized and directional electric field can be applied to the system positioned in this tunnel junction [28, 29]. Generally, the existence of a threshold bias is characteristic of tunneling-electron-induced structural transformation, whereas processes triggered by an electric field (typically in the order of 109 V/m) do not depend on the threshold voltage. In contrast, electric-field-induced processing can act as a "trigger" to induce the structural transition of surface supramolecular assemblies. Nevertheless, in most cases of electric-field-induced processes, the electric field has been induced by changing the bias of imaging or applying a pulse to the STM tip during scanning by the STM tip. Recently, the electric field in an STM tip was used to induce the large-scale reversible transformation of the two distinct assembly phases of a positively charged polycyclic aromatic hydrocarbon [30, 31] and to trigger the formation of physisorbed bilayers [32-35]. Nevertheless, in most cases of electric-field-induced processes, the electric field is induced by changing the bias of imaging or applying a pulse to the STM tip during scanning by the tip. Experimental study of strong electric-field-induced processes at the single-molecule level is still a fairly unchartered territory, and a number of aspects remain unknown. It is especially challenging to search for robust and reproducible systems that can maintain the same response and reversibility after numerous switching cycles.
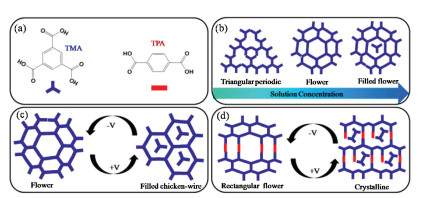
In this contribution, we demonstrate that the large-scale highly ordered triangular fractal framework of TMA can transform into a flower-like structure by increasing its concentration. Then, by reversing the direction of the electric field between the STM tip and highly oriented pyrolytic graphite (HOPG) substrate, the opening and closing of nanoporous structures of TMA on HOPG can be locally controlled. We also propose a porous TMA and terephthalic acid (TPA) bicomponent crystalline network at negative substrate bias can be interchanged in a reversible fashion by simply flipping the polarity of substrate. Based on the experimental results, we suggest the external electric field can be adopted to select the polymorphic outcome of supramolecular assemblies at the solid/liquid interface.
Fig. 1a shows the molecular structures of the TMA and TPA building blocks used in this study. Three different forms of TMA, namely, triangular period, flower-like and filled flower structure, were obtained at a heptanoic acid/HOPG interface by increasing the TMA concentration, as shown in Fig. 1b. Furthermore, when mixed in an appropriate stoichiometric ratio in solution, TMA and TPA formed a mixed self-assembled network, bonded through hydrogen bonding between carboxylic groups of TMA and TPA. We used these perfectly crystalline arrangements as model systems to study the mixing behavior of these molecules under an electric field of varying polarity. Fig. 1c shows the flower-like porous crystalline arrangement changes into honeycomb structure when the substrate bias is reversed from negative to positive. Furthermore, a well-ordered rectangular nanoporous binary flower-like network was destroyed and two TMA molecules appeared in porous when changing the polarity of electric field, as shown in Fig. 1d.

|
Download:
|
| Fig. 1. (a) Molecular structures of TMA and TPA. (b) Schematic showing the TMA concentration dependence of TMA structural frameworks. (c, d) Schematics showing the influence of the surface composition on the outcome of external electric field-induced switching. | |
{kind=link}
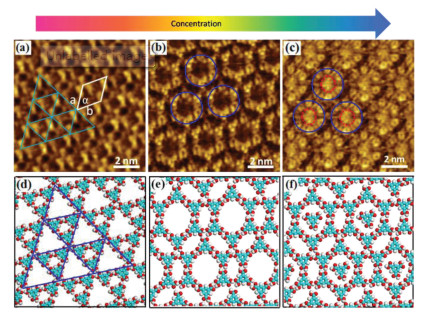
Fig. 2 shows STM images of different host network structures of TMA in heptanoic acid on the surface of HOPG by adjusting the sample concentrations. At a low concentration (2.8 × 10-5 mol/L), we observed the coexistence of a triangular period porous network and a flower-like network, as shown in Fig. S1 (Supporting information). Fig. 2a shows that the triangular fractal porous network was formed by groups of three TMA molecules via hydrogen bonds. Fig. 2d shows the corresponding molecular model with the lattice parameters obtained from calibrated STM images. Interestingly, the triangular period porous network was completely transformed into the flower-like network by an increase in the TMA concentration to 8.2 × 10-5 mol/L (Fig. 2b). It is obvious that one-third of the hydrogen bonds are involved with three TMA molecules, resulting in a circular hydrogen bonding network as presented in Fig. 2e, which is in good agreement with the previous reports [36, 37]. The flower structure is more densely packed (0.99 molecules/nm2) than the triangular period structure (0.89 molecules/nm2). On a further increase in the TMA concentration to 2.5 × 10-4 mol/L, the STM observation in Fig. 2c clearly shows that the basic flower like framework is still observed but now a bright spherical structure was embedded into each pore of the flower-like network. These bright spots actually represent individual TMA molecules. This means that additional TMA molecules are adsorbed on the graphite partially filling all the cavities. Filling of cavities leads however to an increase of the packing density to around 1.15 molecules/nm2. Because TMA is a π-conjugated molecule, the density of the electronic states of the TMA molecules in the pores became very high, forming hydrogen bonds with the six TMA molecules that formed the network pores. The contrast in the STM image is therefore very pronouncing, and the TMA molecules in the pores appear to be larger and brighter than the other TMA molecules. By carefully analyzing the arrangement of the molecules in the STM image, we proposed a model of the self-assembled molecular structure as shown in Fig. 2f. This structure is formed by a similar host-guest mechanism as discussed above for the filled chicken-wire structure [38, 39].

|
Download:
|
| Fig. 2. STM images showing the concentration dependence of the morphology of TMA monolayers at a heptanoic acid/HOPG interface. (a) 2.8 × 10-5 mol/L (I =0.63 nA, V = -0.35 V); (b) 8.2 × 10-5 mol/L (I =0.65 nA, V = -0.32 V); (c) 2.5 × 10-4 mol/L (I =0.60 nA, V = -0.38 V); (d, f) suggested corresponding molecular models of TMA at the respective concentrations. | |
{kind=link}
It was demonstrated previously that at lower concentration more loosely packed and at higher concentration more densely packed structures are favored [16, 40]. To estimate how energetically favorable the formation of a specific polymorph at the interface is, the Gibbs energy has to be considered. At higher concentrations, the mobility of the molecules in the liquid phase increases as well as the diffusion and rotation of the molecules adsorbed on HOPG at the liquid–solid interface, so it easy for TMA molecules to agglomerate and arrange into the corresponding preferential H-bonding motif to reach the thermodynamically favored structure. The kinetically controlled growth of the interfacial monolayer starts when the droplet comes in contact with the substrate and the very first molecules become adsorbed and aggregate. The nucleation process and the following growth of one polymorph are usually in competition with kinetic blockades resulting in domains and in the initial formation of possibly metastable arrangements with a high nucleation and growth rate [41, 42].
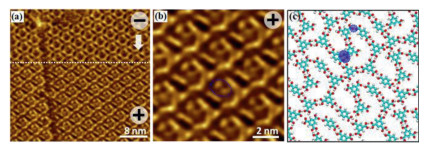
During all the experiments described above, we always carried out measurements at a negative sample bias at room temperature. Surprisingly, we found that the change of the polarity of the electric field applied on the samples led to a structural transition. Fig. 3a illustrates the dependence on the sample bias polarity of the structure of a TMA monolayer physisorbed on a heptanoic acid/HOPG surface. A flower-like monolayer of TMA (8.2 × 10-5 mol/L) was obtained a negative sample bias. The measured unit cell parameters are a = b = 2.5 ± 0.1 nm, and α = 60° ± 1°. The formation of a honeycomb H-bonded network suppresses the TMA deformation and the interaction with the surface, while the stability of the adsorbed network is compensated by H-bonding. When we switched the polarity of the sample bias to positive, we detected bright spherical spots in the honeycomb structure. Six TMA molecules connect to each other by pairs of hydrogen bonds, and a guest TMA molecule is inserted into a hole, shown in Fig. 3b. Fig. 3c shows the corresponding molecular model. Because the cavity area is larger than the size of a single TMA molecule, it can move inside, and it has been observed in various positions forming weak H-bonds to the chicken-wire framework. The honeycomb network of TMA obtained at a negative substrate bias on a nonanoic acid/HOPG surface can also change into a densely packed network that consists of rows of TMA molecules when the substrate bias was reversed. Because TMA is a π-conjugated molecule, the density of the electronic states of the TMA molecules in the pores is very high. Therefore, the contrast in the STM image is very noticeable, and these molecules appear to be larger and brighter than the other TMA molecules. Electric-field-induced structural transitions of the parent TMA network, however, were found to occur almost instantaneously, in that the network changed from one scan line to another upon the reverse of the polarity [43, 44]. Despite its sluggishness in the forward direction, the transition of the network described above was fully reversible and could be initiated by reversing the sample bias to negative.

|
Download:
|
| Fig. 3. (a) STM image showing the reversibility of the phase change of TMA molecules at a heptanoic acid/HOPG interface. (b) High-resolution STM image at a positive substrate bias of the TMA structure at a heptanoic acid/HOPG interface (I =0.47 nA, V = 1.12 V), and six bright spots represent six TMA molecules. (c) Model of the TMA structure at a positive substrate bias. | |
{kind=link}
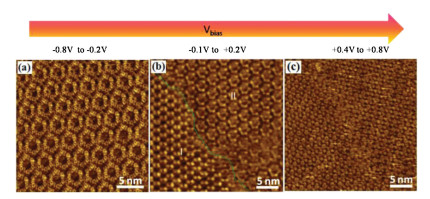
Besides its polarity, the magnitude of the substrate bias also controls the composition of the monolayer phases. A threshold for the observed structural transitions was discovered when the substrate bias was changed gradually in increments of 0.2 V from -0.8 V to +0.8 V. Fig. 4 shows voltage magnitude-dependent structural transitions in the case of TMA. The flower like structure remains stable in the voltage range of -0.8 V to -0.2 V (Fig. 4a). Further changes in the substrate bias lead to formation of chicken-wire and flower coexisting networks within the voltage range of -0.1 V to 0.2 V as shown in Fig. 4b. Additional increase in the field strength furnished the final structure in the filled chicken-wire structure, which remains stable within the voltage window of 0.4 V to 0.8 V (Fig. 4c). The magnitude dependence of the transition can thus alternatively be understood by considering gradual deprotonation of the TMA molecules as a function of increase in the field strength on the positive side of the substrate bias. These systematic measurements reveal that neither of the supramolecular networks observed at positive (+0.8 V) or negative (-0.8 V) bias are stable at or around 0 V when the field is weak or completely removed. The observation of the distorted network indicating partial disruption of regular hydrogen bonding between the two molecules indicates that the partially deprotonated species may be present on the surface at or around zero field.

|
Download:
|
| Fig. 4. Voltage-magnitude-dependent STM image showing the gradual transition of chicken-wire (a) to flower (b) via filled flower (c). Tunneling current (a-c) I =0.62 nA. | |
{kind=link}
TMA is known to assemble multi-dimensional hydrogen bonded host-guest architectures with other molecules consisting carboxyl groups. We chose TPA molecule, with two carboxyl groups in the end of benzene, as the functional guest molecule to modify the network of TMA. TMA and TPA are small rigid molecules, and no dipole moment is induced by the free rotation of their benzene rings. In addition, the carboxyl groups of TMA and TPA formed several in-plane hydrogen bonds via adsorption on the HOPG surface, which means that these hydrogen bonds and the carboxyl groups themselves were coplanar with the rest of the aromatic skeleton. In our previous report, when TMA was mixed with two-fold symmetric TPA with their molar ratio 3:1 at a heptanoic acid/HOPG interface at a negative substrate bias, a well-ordered rectangular nanoporous binary 2D porous flower-like network was formed [45, 46]. Some TMA molecules connected to other three molecules by one-third of hydrogen bonds to form a circular structure. In addition, each TPA forms two hydrogen bonds with one carboxyl group of the neighboring TMA molecules (Fig. S3 in Supporting information). The measured unit cell parameters are a = 2.7 ± 0.1 nm, b = 3.8 ± 0.1 nm, and α = 69° ± 1°. Reversal of the substrate bias led to the appearance of another phase, as shown in Fig. 5a. From the high-resolution STM image in Fig. 5b, it can be clearly observed that the structure of rectangular flower-like network was destroyed. The molecular model illustrated in Fig. 5c reveals that one TMA molecule was removed from the flower-like network, two TMA molecules in nanopores forms one hydrogen bonds with one carboxyl group, and weak aromatic —C—H···O=C— hydrogen bonding occurred between TMA molecules that belonged to the flower-like network. The reverse transition, however, re-accommodated the lost TMA molecules in the network and thereby restored the original rectangular nanoporous binary flower-like network.

|
Download:
|
| Fig. 5. (a) STM image showing the reversibility of the phase change of the TMA/TPA structure at a heptanoic acid/HOPG interface. (b) High-resolution image at a positive substrate bias of the TMA/TPA structure at a heptanoic acid/HOPG interface (I =0.52 nA, V = 0.96 V). (c) Model of the TMA/TPA structure at a positive substrate bias. | |
{kind=link}
The experimental results described above clearly reveal that the networks change from open porous at negative bias to higher packing density structure at positive bias. Under such circumstances, the possible formation of a dipole moment for TMA upon conformational changes proposed by Cometto et al. cannot explain the networks since TMA and TPA both are small rigid planar molecules [47, 48]. Despite their open nature, which does not conform to the thermodynamic principle of close-packing, the stability of such networks is governed by maximization of energy per molecule, in which case each molecule forms six energetically favorable hydrogen bonds with its neighbor. Therefore, not only the overall energy of the supramolecular surface is determined by close packing wherein the energy per unit area dominates, but the carboxylate ion formed after deprotonation may also engage in hydrogen bonding with intact carboxyl groups. A hydrogen bond formed between carboxylate ions and carboxyl groups is known to be extremely strong and is categorized as a low-barrier hydrogen bond. In this case, the hydrogen atom occupies a position approximately equidistant from the two oxygen atoms and is equally shared between the donor and the acceptor. Combining the considerations made above, the following scenario for explaining the switching behavior is brought forward. In agreement with previous studies [49-51], we may conclude that the formation of strong hydrogen bonds between the carboxylate anions and carboxyl groups which may contribute to the overall stabilization of the networks formed at positive substrate bias. This means that the apparent carboxyl-carboxyl contacts in the networks obtained at positive bias could as well be ascribed to carboxyl-carboxylate hydrogen bonding instead of regular R22(8) carboxyl-carboxyl hydrogen bonding. The chemical insensitivity of STM precludes the determination of the exact sites where the plausible deprotonation has occurred.
The triangular period porous network of TMA molecules can be tuned into a flower-like structure by increasing its concentration. Then, by reversing the direction of the electric field that exists between the STM tip and HOPG, the open flower-like nanoporous structure of TMA is stabilized at negative substrate bias, whereas relatively compact networks survive at positive substrate bias. TMA and TPA molecules that form bicomponent crystalline rectangular flower-like network at negative substrate bias are destroyed in a reversible fashion by simply flipping the substrate polarity. In conclusion, structural polymorphs formed at the solution–solid interface can be controlled by adjusting the concentration of the molecules and by changing the sample bias. While a number of mechanistic details still need to be verified experimentally, this work provides a compelling example of electricfield-controlled mixing behavior of surface-adsorbed supramolecular systems.
Declaration of competing interestThe authors declare no competing financial interest.
AcknowledgmentsThis work was supported by Science and Technology Project of Jiangxi Provincial Education Department (Nos. GJJ180942 and GJJ180941); National Natural Science Foundation of China (No. 11664026).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.06.012.
| [1] |
J.V. Barth, G. Costantini, K. Kern, Nature 437 (2005) 671-679. DOI:10.1038/nature04166 |
| [2] |
S. De Feyter, F.C. De Schryver, J. Phys. Chem. B 109 (2005) 4290-4302. DOI:10.1021/jp045298k |
| [3] |
R. Gutzler, T. Sirtl, J.F. Dienstmaier, et al., J. Am. Chem. Soc. 132 (2010) 5084-5090. DOI:10.1021/ja908919r |
| [4] |
Y. Sun, S. Li, Z. Zhou, et al., J. Am. Chem. Soc. 140 (2018) 3257-3263. DOI:10.1021/jacs.7b10769 |
| [5] |
W. Li, J. Jin, X.L. Leng, et al., J. Phys. Chem. C 120 (2016) 12605-12610. DOI:10.1021/acs.jpcc.6b03434 |
| [6] |
M. Lackinger, W.M. Heckl, Langmuir 25 (2009) 11307-11321. DOI:10.1021/la900785f |
| [7] |
G.N. Krishna, I. Oleksandr, A.M. Jill, et al., J. Am. Chem. Soc. 128 (2006) 4212-4213. DOI:10.1021/ja0602896 |
| [8] |
M. Lackinger, S. Griessl, W.M. Heckl, M. Hietschold, G.W. Flynn, Langmuir 21 (2005) 4984-4988. DOI:10.1021/la0467640 |
| [9] |
L. Kampschulte, M. Lackinger, A.K. Maier, et al., J. Phys. Chem. B 110 (2006) 10829-10836. |
| [10] |
J.Q. Li, B. Tu, X.K. Li, et al., Chem. Commun. 55 (2019) 11599-11602. DOI:10.1039/C9CC05872A |
| [11] |
H.Y. Shi, X.C. Lu, Y.H. Liu, et al., ACS Nano 12 (2018) 8781-8790. DOI:10.1021/acsnano.8b05045 |
| [12] |
F. Chen, X. Chen, L.C. Liu, et al., Appl. Phys. Lett. 100 (2012) 081602-081604. DOI:10.1063/1.3685713 |
| [13] |
S. Stepanow, N. Lin, D. Payer, et al., Angew. Chem. Int. Ed. 46 (2007) 710-713. DOI:10.1002/anie.200603644 |
| [14] |
F.F. Xiang, Y. Lu, C. Li, et al., Chem. Eur. J. 21 (2015) 12978-12983. DOI:10.1002/chem.201501434 |
| [15] |
M.O. Blunt, J. Adisoejoso, K. Tahara, et al., J. Am. Chem. Soc. 135 (2013) 12068-12075. DOI:10.1021/ja405585s |
| [16] |
Y.T. Shen, K. Deng, X.M. Zhang, et al., Nano Lett. 11 (2011) 3245-3250. DOI:10.1021/nl201504x |
| [17] |
Z. Chen, J.H. Tang, W.Z. Chen, et al., Organometallics 38 (2019) 4244-4249. DOI:10.1021/acs.organomet.9b00544 |
| [18] |
R. Gutzler, T. Sirtl, J.F. Dienstmaier, et al., J. Am. Chem. Soc. 132 (2010) 5084-5090. DOI:10.1021/ja908919r |
| [19] |
I. Andrius, S. Mantas, J.K. Kasparas, E.T. Evaldas, J. Phys. Chem. C 123 (2019) 3552-3559. DOI:10.1021/acs.jpcc.8b10704 |
| [20] |
H.J. Schneider, Org. Biomol. Chem. 15 (2017) 2146-2151. DOI:10.1039/C6OB02755E |
| [21] |
F. Chiaravalloti, L. Gross, K.H. Rieder, et al., Nat. Mater. 6 (2007) 30-33. DOI:10.1038/nmat1802 |
| [22] |
Q.N. Zheng, X.H. Liu, X.R. Liu, et al., Langmuir 30 (2014) 3034-3040. DOI:10.1021/la5002418 |
| [23] |
J.A.A.W. Elemans, Adv. Funct. Mater. 26 (2016) 8932-8951. DOI:10.1002/adfm.201603145 |
| [24] |
X. Peng, F.Y. Zhao, Y. Peng, J. Li, Q.D. Zeng, Soft Matter 16 (2020) 54-63. DOI:10.1039/C9SM01847F |
| [25] |
T. Kudernac, S.B. Lei, J.A.A.W. Elemans, S. De Feyter, Chem. Soc. Rev. 38 (2009) 402-421. DOI:10.1039/B708902N |
| [26] |
S. Clair, O. Ourdjini, L. Abel, M. Porte, Chem. Commun. 47 (2011) 8028-8030. DOI:10.1039/c1cc12065d |
| [27] |
J. Xu, Y.B. Li, L.J. Wang, et al., Chin. Chem. Lett. 30 (2019) 767-770. DOI:10.1016/j.cclet.2018.09.019 |
| [28] |
G. Velpula, J. Teyssandier, S. De Feyter, K.S. Mali, ACS Nano 11 (2017) 10903-10913. DOI:10.1021/acsnano.7b04610 |
| [29] |
M. Saeed, A. Mahmood, A.S. Saleemi, X.M. Zeng, S.L. Lee, J. Phys. Chem. C 124 (2020) 829-835. DOI:10.1021/acs.jpcc.9b11006 |
| [30] |
K. Cui, K.S. Mali, O. Ivasenko, et al., Angew. Chem. Int. Ed. 53 (2014) 12951-12954. DOI:10.1002/anie.201406246 |
| [31] |
Y. Sun, S. Li, Z. Zhou, et al., J. Am. Chem. Soc. 140 (2018) 3257-3263. DOI:10.1021/jacs.7b10769 |
| [32] |
M. Alemani, M.V. Peters, S. Hecht, et al., J. Am. Chem. Soc. 128 (2006) 14446-14447. DOI:10.1021/ja065449s |
| [33] |
S.B. Lei, K. Deng, Y.L. Yang, et al., Nano Lett. 8 (2008) 1836-1843. DOI:10.1021/nl0803186 |
| [34] |
K.S. Mali, D. Wu, X. Feng, et al., J. Am. Chem. Soc. 133 (2011) 5686-5688. DOI:10.1021/ja111699n |
| [35] |
C. Wang, D. Danovich, H. Chen, S. Shaik, J. Am. Chem. Soc. 141 (2019) 7122-7136. DOI:10.1021/jacs.9b02174 |
| [36] |
A.C. Aragones, N.L. Haworth, N. Darwish, et al., Nature 531 (2016) 88-91. DOI:10.1038/nature16989 |
| [37] |
S.G. Boxer, S.D. Fried, Annu. Rev. Biochem. 86 (2017) 387-415. DOI:10.1146/annurev-biochem-061516-044432 |
| [38] |
S.L. Lee, Y. Fang, G. Velpula, et al., ACS Nano 9 (2015) 11608-11617. DOI:10.1021/acsnano.5b06081 |
| [39] |
S. Griessl, M. Lackinger, M. Edelwirth, M. Hietschold, W.M. Heckl, Single Mol. 3 (2002) 25-31. DOI:10.1002/1438-5171(200204)3:1<25::AID-SIMO25>3.0.CO;2-K |
| [40] |
J.M. Macleod, O. Ivasenko, D.F. Perepichka, F. Rosei, Nanotechnology 18 (2007) 424031-424040. DOI:10.1088/0957-4484/18/42/424031 |
| [41] |
R. Gutzler, L. Cardenas, F. Rosei, Chem. Sci. 2 (2011) 2290-2301. DOI:10.1039/c1sc00531f |
| [42] |
D.C.Y. Nguyen, L. Smykalla, T.N.H. Nguyen, T. Ruffer, M. Hietschold, J. Phys. Chem. C 120 (2016) 11027-11036. DOI:10.1021/acs.jpcc.6b03409 |
| [43] |
S.L. Lee, Y.J. Hsu, H.J. Wu, et al., Chem. Commun. 48 (2012) 11748-11750. DOI:10.1039/c2cc35413f |
| [44] |
Y.P. Mo, T. Chen, J.X. Dai, K. Wu, D. Wang, J. Am. Chem. Soc. 141 (2019) 11378-11382. DOI:10.1021/jacs.9b04815 |
| [45] |
V.V. Korolkov, S. Allen, C.J. Roberts, S.J.B. Tendler, J. Phys. Chem. C 116 (2012) 11519-11525. |
| [46] |
W. Li, X.L. Leng, C.Y. Xu, N. Liu, Phys. E: Low. Syst. Nanostruct. 101 (2018) 197-200. DOI:10.1016/j.physe.2018.04.013 |
| [47] |
F.P. Cometto, K. Kern, M. Lingenfelder, ACS Nao 9 (2015) 5544-5550. DOI:10.1021/acsnano.5b01658 |
| [48] |
F. Shayeganfar, A. Rochefort, Langmuir 30 (2014) 9707-9716. DOI:10.1021/la501619b |
| [49] |
A. Mahmood, M. Saeed, Y. Chan, et al., Langmuir 35 (2019) 8031-8037. DOI:10.1021/acs.langmuir.9b00569 |
| [50] |
J. Ubink, M. Enache, M. Stohr, J. Chem. Phys. 148 (2018). |
| [51] |
S.Y. Li, X.Q. Yang, T. Chen, et al., ACS Nano 13 (2019) 6751-6759. DOI:10.1021/acsnano.9b01337 |