 2021, Vol. 32
2021, Vol. 32 


b Department of Chemistry, Hunan University of Science and Engineering, Yongzhou 425100, China;
c Hunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, Changsha University of Science and Technology, Changsha 410114, China;
d School of Chemistry and Chemical Engineering, University of South China, Hengyang 421001, China
"Preventing waste" is the first and most basic of the 12 principles of green chemistry. The principle states that compared with the troublesome treatment or cleaning after the waste generated, the prevention of waste generation is much more advantageous. Nowadays, the vast majority of the fine chemicals and pharmaceuticals are manufactured via homogeneous catalytic processes. However, the homogeneous catalyst and solvent are often difficult to be reused and the disposal of them often causes environmental concerns. In addition, the expected products from reaction mixtures are often mixed with un-reacted reagent and undesired byproducts, making it necessary to perform tedious isolation and purification procedures. Such treatment process pushes up production costs, pollutes the environment, and also limits the application of the one-pot method, hindering subsequent conversion of products. As a consequence, the development of novel homogeneous catalytic reactions in which both the catalyst and solvent could be re-used directly and the products could be collected via material- and energy-consuming isolation and purification procedures is strongly desirable and presents a considerable challenge in fine chemical industry and pharmaceutical industry [1].
As a potent privileged structure unit in drug research and development, the uracil skeleton is the core structure of many natural products and biologically active molecules exhibiting diverse activities [2]. It has been proved that the purposeful introduction of selenium groups into the molecular structure can improve the physical and chemical properties of organic molecules [3]. Among the uracil derivatives, 5-organylselanyl uracil is a particularly important one, and it has wide applications as part of anti-cancer drugs and anti-viral drugs [4]. Several methods have been developed to prepare these compounds [5]. Given the abundance and accessibility of diorganyl diselenides [6], the direct synthesis of 5-organylselanyl uracils from diselenides represents an attractive alternative. In 1995, Kim and co-workers developed the Mn(OAc)3·2H2O-mediated oxidative cross-coupling of unprotected uracils and diselenides (1.5 equiv.) in AcOH at 70 ℃ to give 5-organylselanyl uracils, but only one example (5-phenylselanyluracil) was presented (Scheme 1a) [5c]. Recently, Cheng reported a method for the preparation of 5-organylselanyl uracils via the NaI/H2O2-mediated selenylation of unprotected uracils with diorganyl diselenides (1.5 equiv.) in DMSO (Scheme 1b) [5d]. However, practical applications of the above-mentioned homogeneous synthetic methods are largely limited to the higher cost of large excess of reagents and the difficult collection of the target products from large amount of un-consumed reagents and un-expected byproducts. To satisfy the needs for 5-organylselanyl uracils in pharmaceuticals, it is highly desirable to develop a cost-effective, sustainable and practical method for synthesizing such molecules.

|
Download:
|
| Scheme 1. Synthesis of 5-organylselanyluracils. | |
Organic electrochemical synthesis can avoid harmful oxidants by using current for red-ox reactions [7]. The current is trace-free and can come from renewable energy sources, such as solar or wind energy, which helps to increase the sustainability of the reaction. Under the guidance of green chemistry principles [8], we herein disclose a sustainable and practical synthetic protocol for various 5-organylselanyl uracils via electrochemical (the 6th Principle) selenylation of uracils with diselenides (the 2nd Principle) at ambient temperature under oxidant-free (the 4th Principle) and external electrolyte-free conditions (Scheme 1c).
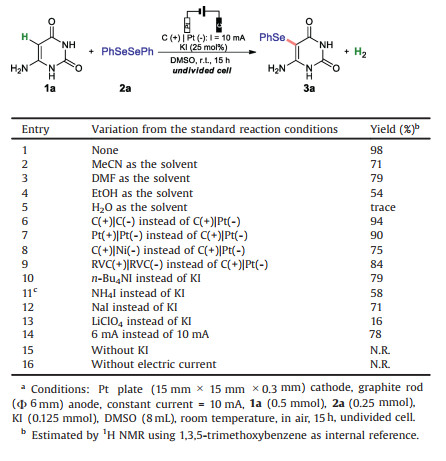
At the outset of our investigations, 6-aminouracil (1a) and diphenyl diselenide (2a) were selected as the template substrates to screen the reaction conditions (Table 1). Employing KI as the catalyst, and DMSO as the reaction solvent, the target product 3a formed in 98% NMR yield under 10 mA constant current with a platinum plate (Pt-) as the anode, a graphite rod (C+) as the cathode at ambient temperature (Table 1, entry 1). Replacing DMSO with MeCN, DMF or EtOH delivered a decreased yield, while only a trace amount of 3a was detected when employing pure water as the reaction medium (entries 2–5). The effect of different electrode materials on reaction efficiency was subsequently explored (entries 6–9), and the experimental results suggested that the platinum plate anode and graphite rod cathode were the most suitable electrode materials in the present reaction. When other iodides, such as n-Bu4NI or NH4I or NaI, were used instead of KI, the yield of the 3a was decreased (entries 10–12). By changing KI to LiClO4, the yield of 3a dropped down to 16% (entry 13). Decreasing the constant current from 10 mA to 6 mA resulted in a lower yield of 3a (entry 14). As a control experiment, no reaction was detected when the selenylation reaction was carried out in the absence of KI or constant current (entries 15 and 16).
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
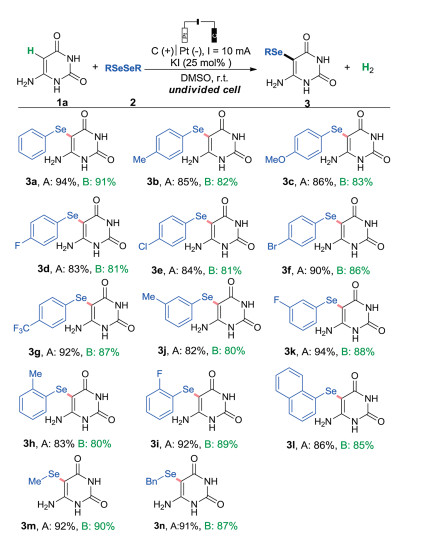
Having established optimal reaction conditions (Table 1, entry 1), the substrate scope of the electrochemical selenylation reaction was next investigated with respect to both diorganyl diselenides and uracils. As illustrated in Scheme 2, all the examined diselenides showed high reactivity delivering the selenylated products in excellent yields. Neither the electronic effect nor steric hindrance effects of the substituent group in phenyl ring of diphenyl diselenides had a considerable influence on the present electrochemical transformation (3a-3i). A bulky 1,2-di(naphthalen-2-yl)diselane substrate was also effective to generate the target product (3l) in 86% yield. Aliphatic diselenides were efficiently converted into the corresponding products (3m) and (3n) in 92% and 91% yields, respectively. In sharp contrast with that an excess of diselenides was needed in the previous reported method, the current reaction employed equimolar amounts of reactants, thus avoiding large amounts of residues of diselenides and un-desired byproducts. Significantly, all the selenylated products were synthesized with high yields and the pure products were collected through water precipitation method.

|
Download:
|
| Scheme 2. Reaction scope of diorganyldiselenide. Conditions: Pt plate (15 mm × 15 mm × 0.3 mm) cathode, graphite rod anode (Φ 6 mm), constant current = 10 mA, 1a (0.5 mmol), 2a (0.25 mmol), KI (0.125 mmol), DMSO (8 mL), room temperature, in air, undivided cell. A: Isolated yields via flash chromatography. B: Isolated yields via water precipitation method. | |
{kind=link}
Next, we tested the scope of the uracil substrates (Scheme 3). An exquisite regioselectivity profile was found for a wide range of unsubstituted uracils and 5-organylselanyl uracils were exclusively formed in excellent yields (3o-3u). A series of uracil derivatives bearing electron-rich or electron-deficient substituents on the pyrimidine ring reacted smoothly resulting in the desired products in good to high yields (3v–3ad). The present protocol is also applicable for the synthesis of 5-organylselanyl thiouracil (3ae).

|
Download:
|
| Scheme 3. Reaction scope of uracils. Conditions: Pt plate (15 mm × 15 mm × 0.3 mm) cathode, graphite rod anode (Φ 6 mm), constant current = 10 mA, 1a (0.5 mmol), 2a (0.25 mmol), KI (0.125 mmol), DMSO (8 mL), room temperature, in air, undivided cell. Isolated yields via flash chromatography. | |
{kind=link}
As a practical synthetic protocol, both the scalability and operational simplicity have great significance for the manufacture of functionalized uracils in laboratory, and even in the chemistry and pharmaceutical industry. Therefore, both large-scale preparation and one-pot transformations from commercially available uracils were carried out. For the 0.5 mmol scale selenylation reaction, decreasing the loading of KI led to a low yield of 3a. Pleasingly, the usage of 20 mol% catalyst could deliver product 3a in 93% yield with the increase of the present reaction scale by 20-fold (Scheme 4). Moreover, an 8-fold increase of the uracil substrate concentration was also feasible, thus minimizing the usage amount of solvent. Importantly, high-purity 3a can be simply collected by water precipitation without the usage of any organic solvent (Fig. 1). The in-vitro cytoxicity of compound 3a was investigated by employing methylthiazoltetrazolium assay on human cervical cancer cells (HeLa), with 5-fluorouracil (a well-known anticancer drug) as the positive control. We are pleased to find that compound 3a exhibits strong cytotoxicity against HeLa with the IC50 = 13.39 mg/mL (See Supporting information). The in-depth research of the pharmacological activitives of 5-organylselanyl uracils is being studied in our laboratory.

|
Download:
|
| Scheme 4. Large-scale synthesis of 3a. | |
{kind=link}

|
Download:
|
| Fig. 1. Self-separating system. (A) before reaction; (B) after reaction; (C) phase after added water; (D) dry product. | |
{kind=link}
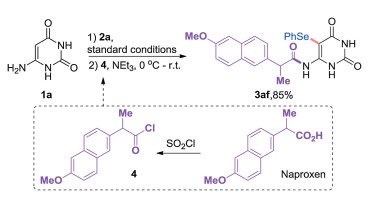
Since both tedious post-processing and purification were need for the previous works to remove the excess amount of reagents and byproducts that will restrain the directly subsequent transformations, the present selenylation reaction has obvious advantages for its excellent atom economy, and it only generates the harmless hydrogen as the sole byproduct. In order to further prove the advantage of the current method, a one-pot transformation starting from substrate 1a was carried out. Naproxen, a famous nonsteroidal anti-inflammatory drug, its corresponding uracil analogue was easily obtained in excellent yield through one-pot sequential selenylation and amidation from unprotected 6-aminouracil (1a → 3af, Scheme 5).

|
Download:
|
| Scheme 5. One-pot transformation. | |
{kind=link}
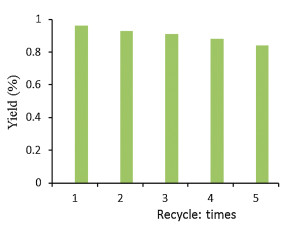
It is well known that the reusability of catalytic system is of great importance from the views of industry and green chemistry. As a consequence, the reusability of the electrochemical system was next investigated by the template reaction using substrate 1a (5 mmol) under the optimal reaction conditions. After completion of the first selenylation reaction, the product 3a was collected via water precipitation and simple filtration. The KI/DMSO filtrate was re-used directly after drying under vacuum. As depicted in Fig. 2, the KI/DMSO system could be facilely re-used, and no significant reduction in activity has been observed after five consecutive runs.

|
Download:
|
| Fig. 2. The reusability of catalytic reaction. | |
{kind=link}
To evaluate the environmental friendliness of the present electrochemical reaction, both the atom economy and Eco-scale value [9] were calculated (Scheme 6). The present method not only presented 2.7-fold increase in atom economy and 2.1-fold increase in Eco-scale value, but also greatly simplified the process for isolating and purifying.

|
Download:
|
| Scheme 6. Comparison of green metrics. | |
{kind=link}
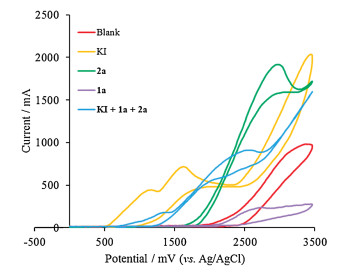
To elucidate the mechanism of the present electrochemical selenylation reaction, some control experiments were conducted. When 2 equiv. of TEMPO (a well-known radical scavenger) were added to the reaction system, the selenylation reaction proceeded as normal, indicating that the free-radical is not involved in the current reaction (Scheme 7a). Performing the reaction under nitrogen atmosphere afforded the product 3a in 95% NMR yield (Scheme 7b). This result indicated the current system is not associated with ambient molecular oxygen. Cyclic voltammetric analysis was also carried out to understand the possible mechanism. As shown in Fig. 3, uracil (1a) could not be oxidized up to 3.5 V, whereas diselenide (2a) was found to be oxidized at 2.97 V (vs. Ag/AgCl). In addition, KI gave two obvious oxidation peaks at 1.15 V and 1.62 V (vs. Ag/AgCl), which indicated that the iodide ion was easier to oxidize at the surface of the anode to form iodine cation, which promoted the formation of oxidation state of diselenide (2a). The similar results were also can be observed in the cyclic voltammetry experiment of reaction mixture of KI, uracil (1a) and diselenide (2a).

|
Download:
|
| Scheme 7. Control experiments. | |
{kind=link}

|
Download:
|
| Fig. 3. The cyclic voltammetry experiment. | |
{kind=link}
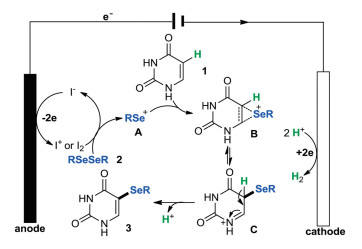
According to the above observations and relevant literature reports [5d], a plausible reaction mechanism for the current reaction is proposed in Scheme 8. Firstly, the anodic oxidation of an iodide ion could produce the iodine cation (I+) or molecular iodine (I2), which could react with a diselenide (2) to generate the highly reactive selenium cation intermediate A. Next, the electrophilic addition of uracil (1) and intermediate A led to a seleranium cation B and/or iminium ion C. Last, the protonation of intermediate C afforded the target product 3. The cathodic reduction of proton produced the hydrogen gas as the sole byproduct. We attempted to detect hydrogen by GC–MS, but all efforts failed.

|
Download:
|
| Scheme 8. Plausible mechanism. | |
{kind=link}
In conclusion, we have presented an eco-friendly, sustainable and practical method for the synthesis of 5-organylselanyl uracils (31 examples, 77%–94%) via the electrochemical selenylation of uracils and diorganyl diselenides at room temperature under oxidant- and external electrolyte-free conditions. The pure products could be obtained through water precipitation and simple filtration, thus avoiding environmentally unfriendly isolation and purification procedures. Other characteristics of this present transformation are as follows: (a) The employment of equal amount of reactants, the usage of electric current as a traceless oxidant, and the generation of hydrogen gas as the sole byproduct greatly simplified the selenylation reaction, thus the one-pot sequential transformation starting from uracil was successfully achieved; (b) Compared with the existing procedure, the present reaction has 2.7-fold increase in atom economy and 2.1-fold increase in Eco-scale value. Given the easy availability of starting materials, the safe and mild conditions, and operational simplicity, this developed method is expected to be ideal for fine chemical industry and pharmaceutical industry.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe are grateful for financial support from the National Natural Science Foundation of China (No. 21902014), the Basic Frontier Research Project of Chongqing (No. Cstc2018jcyjAX0051), and Hunan Provincial Natural Science Foundation of China (No. 2019JJ20008).
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.034.
| [1] |
(a) Y. Kim, C.J. Li, Green Synth. Catal. 1 (2020) 1-11; (b) Z. Cao, Q. Zhu, Y.W. Lin, W.M. He, Chin. Chem. Lett. 30 (2019) 2132-2138; (c) K.J. Liu, T.Y. Zeng, J.L. Zeng, et al., Chin. Chem. Lett. 30 (2019) 2304-2308; (d) J. Shi, W. Wei, Chin. J. Org. Chem. 40 (2020) 2170-2172; (e) L.Y. Xie, Y.S. Liu, H.R. Ding, et al., Chin. J. Catal. 41 (2020) 1168-1173; (f) J.Y. Liao, Q.Y. Wu, X. Lu, et al., Green Chem. 21 (2019) 6567-6573. |
| [2] |
(a) A. Pałasz, D. Ciez, Eur. J. Med. Chem. 97 (2015) 582-611; (b) D.P. Angelika, D. Joanna, P. Marlena, et al., Anticancer Agents Med. Chem. 20 (2020) 359-368. |
| [3] |
(a) B. Banerjee, M. Koketsu, Coord. Chem. Rev. 339 (2017) 104-127; (b) Q.B. Zhang, Y.L. Ban, P.F. Yuan, et al., Green Chem. 19 (2017) 5559-5563; (c) T. Guo, X.N. Wei, Y. Liu, P.K. Zhang, Y.H. Zhao, Org. Chem. Front. 6 (2019) 1414-1422; (d) P. Ni, J. Tan, W. Zhao, et al., Org. Lett. 21 (2019) 3518-3522; (e) T. Guo, X.N. Wei, M. Zhang, et al., Chem. Commun. 56 (2020) 5751-5754; (f) X. Xu, D. Chen, Z. Wang, Chin. J. Org. Chem. 39 (2020) 3338-3352; (g) J. Zhang, H. Wang, Y. Chen, et al., Chin. Chem. Lett. 31 (2020) 1576-1579; (h) P. Ni, J. Tan, W. Zhao, H. Huang, G.J. Deng, Adv. Synth. Catal. 361 (2019) 5351-5356. |
| [4] |
(a) M. Sosnowska, S. Makurat, M. Zdrowowicz, J. Rak, J. Phys. Chem. B 121 (2017) 6139-6147; (b) D. Bartolini, L. Sancineto, A. Fabro de Bem, et al., Selenocompounds in cancer therapy: an overview, in: K.D. Tew, F. Galli (Eds.), Advances in Cancer Research, Vol. 136, Academic Press, 2017, pp. 259-302; (c) S. Makurat, M. Zdrowowicz, L. Chomicz-Mańka, et al., RSC Adv. 8 (2018) 21378-21388. |
| [5] |
(a) K. Anzai, J. Heterocycl. Chem. 16 (1979) 567-569; (b) A. Jana, A.K. Panday, R. Mishra, T. Parvin, L.H. Choudhury, ChemistrySelect 2 (2017) 9420-9424; (c) D.H. Lee, Y.H. Kim, Synlett 1995 (1995) 349-350; (d) X.D. Li, Y.T. Gao, Y.J. Sun, et al., Org. Lett. 21 (2019) 6643-6647. |
| [6] |
(a) K. Sun, X. Wang, F. Fu, et al., Green Chem. 19 (2017) 1490-1493; (b) W. Wei, H. Cui, H. Yue, D. Yang, Green Chem. 20 (2018) 3197-3202; (c) K. Sun, Z. Shi, Z. Liu, et al., Org. Lett. 20 (2018) 6687-6690; (d) D. Yang, G. Li, C. Xing, et al., Org. Chem. Front. 5 (2018) 2974-2979; (e) J. Hua, Z. Fang, J. Xu, et al., Green Chem. 21 (2019) 4706-4711; (f) K. Sun, S. Wang, R. Feng, et al., Org. Lett. 21 (2019) 2052-2055; (g) X.L. Ma, Q. Wang, X.Y. Feng, et al., Green Chem. 21 (2019) 3547-3551; (h) B. Huang, Y. Li, C. Yang, W. Xia, Green Chem. 22 (2020) 2804-2809; (i) J. Hua, Z. Fang, M. Bian, et al., ChemSusChem 13 (2020) 2053-2059. |
| [7] |
(a) J. Wen, L. Zhang, X. Yang, et al., Green Chem. 21 (2019) 3597-3601; (b) S. Zhang, L. Li, J. Zhang, et al., Chem. Sci. 10 (2019) 3181-3185; (c) H. Wang, J. Shi, J. Tan, et al., Org. Lett. 21 (2019) 9430-9433; (d) X. Kong, Y. Liu, L. Lin, Q. Chen, B. Xu, Green Chem. 21 (2019) 3796-3801; (e) J. Zhou, X.Z. Tao, J.J. Dai, et al., Chem. Commun. 55 (2019) 9208-9211; (f) C. Wan, R.J. Song, J.H. Li, Org. Lett. 21 (2019) 2800-2803; (g) G.Y. Dou, Y.Y. Jiang, K. Xu, C.C. Zeng, Org. Chem. Front. 6 (2019) 2392-2397; (h) Y.Z. Yang, R.J. Song, J.H. Li, Org. Lett. 21 (2019) 3228-3231; (i) Z. Zhang, J. Yan, D. Ma, J. Sun, Chin. Chem. Lett. 30 (2019) 1509-1511; (j) H. Ding, K. Xu, C.C. Zeng, J. Catal. 381 (2020) 38-43; (k) J.Y. Chen, H.Y. Wu, Q.W. Gui, et al., Org. Lett. 22 (2020) 2206-2209; (l) X. Meng, Y. Zhang, J. Luo, et al., Org. Lett. 22 (2020) 1169-1174; (m) M.X. He, Z.Y. Mo, Z.Q. Wang, et al., Org. Lett. 22 (2020) 724-728; (n) L.B. Zhang, R.S. Geng, Z.C. Wang, et al., Green Chem. 22 (2020) 16-21; (o) T.S. Zhang, W.J. Hao, R. Wang, et al., Green Chem. 22 (2020) 4259-4269; (p) J. Zhong, Y. Yu, D. Zhang, K. Ye, Chin. Chem. Lett. (2020), doi: http://dx.doi.org/10.1016/j.cclet.2020.08.011. |
| [8] |
(a) L.H. Lu, Z. Wang, W. Xia, et al., Chin. Chem. Lett. 30 (2019) 1237-1240; (b) L.Y. Xie, T.G. Fang, J.X. Tan, et al., Green Chem. 21 (2019) 3858-3863; (c) W.H. Bao, Z. Wang, X. Tang, et al., Chin. Chem. Lett. 30 (2019) 2259-2262; (d) W.B. He, L.Q. Gao, X.J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1895-1898; (e) K.J. Liu, J.H. Deng, T.Y. Zeng, et al., Chin. Chem. Lett. 31 (2020) 1868-1872; (f) L.Y. Xie, Y.S. Bai, X.Q. Xu, et al., Green Chem. 22 (2020) 1720-1725; (g) L.Y. Xie, S. Peng, T.G. Fan, et al., Sci. China Chem. 62 (2019) 460-464. |
| [9] |
K. Van Aken, L. Strekowski, L. Patiny, Beilstein J. Org. Chem. 2 (2006) 3-9. |