 2021, Vol. 32
2021, Vol. 32 


In the past several decades, significant progress has been made in the formation of C—C bonds through the direct functionalization of C—H bonds catalyzed by homogeneous transition-metal catalyst [1]. Transition metal catalysts, in particular palladium, have become a powerful strategy to directly access complex molecules [2]. Compared with a great deal of C(sp2)-H functionalization reactions, selective C(sp3)-H functionalization poses a unique set of challenges due to its additional conformational degrees of freedom and the absence of stabilizing π−orbital interactions with the metal center [3]. Several strategies for C(sp3)–H functionalization have been described using an adjacent directing group (DG) to locate a catalyst in an appropriate spatial arrangement [4]. Through coordination of a directing group to palladium, the proximity effect can often override the inherent steric/electronic preferences and ultimately realize the site-selectivity of C(sp3)–H bond functionalization. But this strategy requires the additional two steps for their stoichiometric installation and removal of the directing ligands on substrates which reduce the overall efficiency of the reaction process (Scheme 1a).

|
Download:
|
| Scheme 1. The development of C(sp3)-H arylation. | |
Very recently, the concept of transient directing groups (TDG) has emerged. During the reaction, TDG is covalently bound to the substrate, coordinated with the metal center for site-selective C(sp3)-H functionalization, and finally released, the product was formed simultaneously (Scheme 1b) [5]. Catalyzed by palladium, Yu [6], Hu [7] and Ge's groups [8] described C(sp3)–H arylation of aldehydes or ketones with α-amino acids, acetylhydrazone and β-amino acid as TDG, respectively. One thing all these reports have in common is using iodobenzenes as arylation reagents and transient imines as 'next generation' directing groups.
Considering the expensive price and high toxicity of palladium catalyst, heterogeneous palladium catalyst coordinated with transient directing strategy is a highly attractive protocol owing to its chemical economic and environmentally benign fashion. In recent years, higher order Pd species-supported palladium nanoparticles (PdNPs) has proven to be a highly selective and efficient heterogeneous catalyst for C–H bond functionalization reactions [9]. Our previous work has also confirmed that the Pd(OAc)2 could be replaced by Pd0/γ-Al2O3 catalyst in C(sp2)-H functionalization reactions [10]. But aiming to accomplish the heterogeneous palladium catalyzed C(sp3)-H functionalization reaction using a TDG, two basic problems should be solved: 1) Considering the catalytic mechanism (via Pd0/PdⅡ/PdⅣ or PdⅡ/PdⅣ catalytic cycle), which valence supported PdNPs catalyst is proper to the reaction; 2) Another fundamental obstacle is that the superstoichiometric quantities of Ag salt were necessary to abstract iodide of iodobenzenes [11] which will generate a great deal of AgI precipitate, leading to the hard recovery of catalyst by simple precipitation method.
Herein, we reported, for the first time, a supported metallic state PdNPs catalyzed C(sp3)-H bonds arylation by a transient directing strategy (Scheme 1c). The PdNPs supported on four various oxide powders, including -Al2O3, ZnO, Sm2O3 and NiO, were prepared by the modified impregnation-reduction method [10] (details are deposited in Supporting information) and applied to the C(sp3)–H bond arylation of 2-methylbenzaldehyde 1a with 4-iodoanisole 2a using acetohydrazide as TDG, AgTFA as oxidant. After 48 h of refluxing in AcOH-HFIP (v/v, 3:1) in the presence of 3 wt% Pd/γ-Al2O3, 3 wt% Pd/ZnO, 3 wt% Pd/Sm2O3 and 3% Pd/NiO, the mono-arylation product 3aa was isolated in yields of 75%, 33%, 50% and 69%, respectively (Table S1 in Supporting information). This result clearly indicated that the heterogeneous catalysis by a transient directing strategy was a feasible route. The higher activity of PdNPs on γ-Al2O3 was for the reason that γ-Al2O3 had a large surface area and open porosity which could enable a high dispersion of the PdNP catalyst [12]. Compared with our catalyst, commercial 5 wt% Pd/C showed lower catalytic performance. Other TDG, such as glycine, l-alanine, l-valine, and 2-aminoisobutyric acid, showed much lower activity for this heterogeneous catalytic coupling reaction. A control experiment was carried out in the absence of TDG, which gave no product, suggesting TDG is vital in this reaction (details are deposited in Supporting information).
|
|
Table 1 The scope of aryl iodides.a |

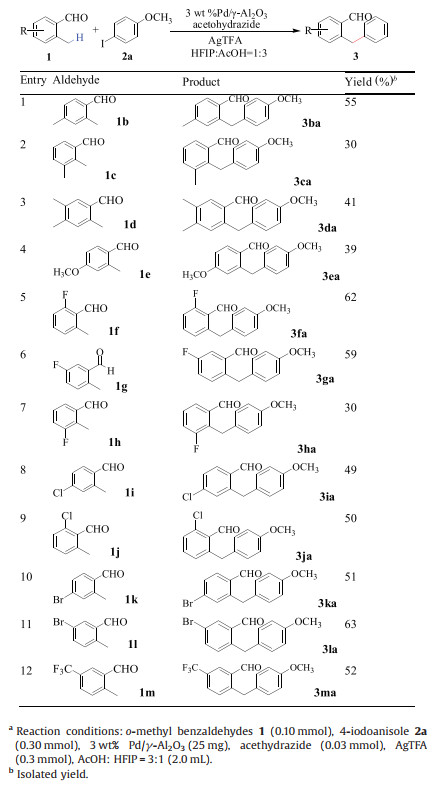
With optimized conditions in hand, the scope of aryl iodides was first studied. As shown in Table 1, para-, meta- and ortho- substituted substrates were chosen to examine the effect of steric hindrance on this reaction. The results clearly indicated that both para- and ortho-substituted aryl iodides (2a with 2b) could obtain the corresponding products in similar yields. There is also no clear difference in yields for the para- and meta-substituted aryl iodides (2h and 2i). These results suggested that steric hindrance could not significantly affect the reaction. The reaction can tolerate various functional groups, including alkoxy, alkyl, halogen (F, Br and Cl), acyl, ester, trifluoromethyl, and nitro group. Notably, bromo-substituted aryl iodides afforded the corresponding products in the best yield, preserving the halogen groups for further synthetic elaborations via cross coupling reactions.
The scope of o-methylbenzaldehydes with different substituent groups was next explored using 4-iodoanisole 2a as the arylation reagent (Table 2). o-Methylbenzaldehydes bearing substituents at all the positions of benzene ring underwent C–H arylation with 2a to provide the products in moderate yields. However, the use of a sterically hindered 3-substituted o-methylbenzaldehydes led to a reduced yield (entries 2 and 7). These results indicated that steric hindrance of o-methylbenzaldehydes could significantly affect this reaction. Both electron-withdrawing (1b-1e) and electron-donating substituents (1f-1m) were tolerated in the heterogeneous C(sp3)-H activation reaction, and their reactivities did not show a significant difference.
|
|
Table 2 The scope of o-methylbenzaldehydes with different substituent groups.a |
To investigate the practical application of this newly heterogeneous catalytic reaction in organic synthesis, we conducted a gram-scale reaction of 1a (1 g) with 2a in the presence of 3 wt% Pd/γ-Al2O3 catalyst, and isolated the desired product 3aa in 70% yield (Experimental section in Supporting information). As we can see, even though the reaction scale was magnified up to 83 times, ideal synthetically yields still could be obtained.
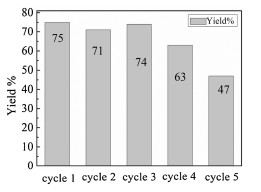
We also examined the possibility of recycling the PdNPs catalyst in the reaction of o-methylbenzaldehyde 1a with 4-iodoanisole 2a. After each reaction cycle, the centrifugated deposit was washed with ammonia aqueous to get rid of AgI precipitate. As shown in Fig. 1, although the catalytic performance of the PdNPs did not declined apparently after 3rd recycle, but the yield of desired product dived to 47% in 5th recycle.

|
Download:
|
| Fig. 1. Recyclability of the catalyst. | |
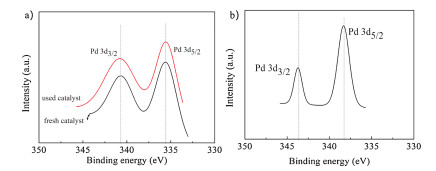
In order to get valence state information of Pd on the catalyst surface, the fresh and used (after 1st recycle and after 5th recycle) 3 wt% Pd/γ-Al2O3 catalysts were tested by XPS analysis (Fig. 2). It is showed that PdNPs on the support consisted of metallic Pd, as indicated by the Pd0 peak at a binding energy for 335.54 eV of Pd 3d5/2 and 340.76 eV of Pd 3d3/2 over fresh catalyst, 335.58 eV of Pd 3d5/2 and 340.82 eV of Pd 3d3/2 over used catalyst [13]. There was no significant change in the quantity of the active Pd0 species in the Pd/γ-Al2O3 catalyst before and after the reactions. Hence, the small decrease in the isolated yield of the arylation product in the recycling experiments was probably not due to deactivation of the active catalytic species, but attributed to some loss of the nanomaterial in the recovery process. Meanwhile, this result indicated that the PdNPs-catalyzed C(sp3)-H bonds functionalization reaction was performed via a catalytic cycle that began with Pd0. The inference is same as Hu's proposed mechanism [7]. But the XPS signal appeared at binding energies for 338.37 eV of Pd 3d5/2 and 343.76 eV of Pd 3d3/2 over after 5th recycle catalyst. This result indicated that the metallic state palladium was fully oxidized to PdⅡ species and it will inevitably affect the catalytic performance of nano-catalyst.
The amounts of Pd loading in the samples were determined by atomic absorption spectrophotometer (AAS), and the Pd content of the fresh catalyst is approximately 3 wt% (Table 3). The used catalyst cycled one times had almost same Pd loading (2.96 wt%) compared with the fresh catalyst.
|
|
Table 3 Characterization results of AAS of catalysts. |

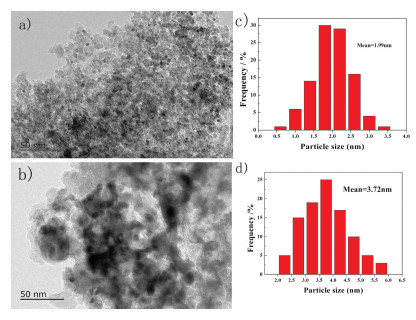
The transmission electron microscopic (TEM) pictures of the fresh and used (after 1st recycle) 3 wt% Pd/γ-Al2O3 catalysts are given in Fig. 3. The PdNPs of fresh and used catalysts were both well dispersed and had narrow size distributions. The Pd particle size was measured and found to be 1.99 nm and 3.72 nm for fresh and used catalysts, respectively. The used catalyst had a larger Pd particle size but PdNPs still distributed evenly on the γ-Al2O3 surface, and no apparent agglomeration was observed.

|
Download:
|
| Fig. 3. TEM images of 3 wt% Pd/γ-Al2O3: (a) fresh catalyst; (b) used (after 1st recycle) catalyst; (c, d) The size distributions of fresh and used catalysts were obtained from TEM images by counting > 200 isolated palladium particles in the images of a sample. | |
We have carried out two experiments to determine the heterogeneity of the catalyst. At first, we removed the catalyst by filtration over Celite from the model reaction and measured the palladium content in solution by ICP-MS. The palladium content of the reaction mixture was found to be 2.0 ppm, demonstrating that trace Pd metal on the solid supported surface leached into the reaction medium. Then, to demonstrate that the trace palladium in the reaction mixture is not the source of catalytic activity, a hot filtration test of the model reaction mixture was executed and GC data are given in Table 4. The results indicated that the substrate turnover ceases after filtration. This is intended to preclude the possibility of homogeneous catalysis. Overall, the PdNPs catalyzed C(sp3)-H activation have the advantage over homogeneous methods, which requires additional steps to furnish the desired product.
|
|
Table 4 The results of hot filtration test. |

{kind=link}
{kind=link}
{kind=link}
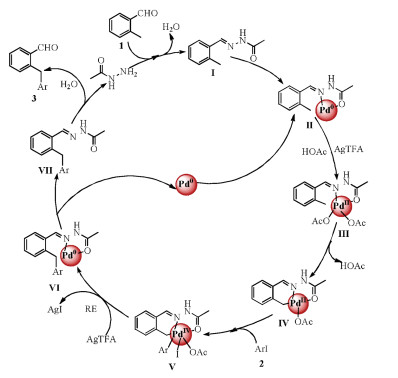
Based on the commonly accepted mechanism from the literature, the proposed reaction pathway is shown in Scheme 2. Initially, aldehyde reacts with acetohydrazide to form acetohydrazone Ⅰ, which serves as a directing group in the next step. Then, bidentate coordination of the acetohydrazone moiety in Ⅰ to the supported palladium nanoparticles occurs to form the chelated cyclometalated Pd-complex Ⅱ. Then AgTFA/AcOH-promoted oxidation of Pd0 into the intermediate Ⅲ [7], and subsequently, PdⅡ activates the ortho C(sp3)-H bond to generate cyclopalladated intermediate Ⅳ and HOAc. Then, oxidative addition of intermediate Ⅳ and aryl iodide takes place to form intermediate Ⅴ [14]. The iodide of intermediate Ⅴ was captured by AgTFA to generate AgI precipitate and then it undergoes reductive elimination to obtain intermediate Ⅵ. Subsequently, intermediate Ⅵ is decomposed to intermediate Ⅶ along with the Pd0 species to complete the catalytic cycle [7]. Intermediate Ⅶ is hydrolyzed to the desired product 3 and acetohydrazide.

|
Download:
|
| Scheme 2. Proposed mechanism. | |
{kind=link}

|
Download:
|
| Fig. 2. (a) XPS spectra of fresh and used (after 1st recycle) 3 wt% Pd/γ-Al2O3; (b) XPS spectra of used (after 5th recycle) 3 wt% Pd/γ-Al2O3. | |
{kind=link}
In summary, it was found for the first time that heterogeneous palladium catalyzed C(sp3)-H bonds functionalization reaction by a transient directing strategy. Using 3 wt% Pd/γ-Al2O3 as heterogeneous catalyst and acetohydrazide as TDG, a broad scope of o-methylbenzaldehydes react with various para-, meta-, and ortho-substituted aryl iodides to synthesize the corresponding mono-arylation product in yields up to 84%. Also, the reaction can tolerate various functional groups, including alkoxy, alkyl, halogen (F, Br and Cl), acyl, ester, trifluoromethyl, and nitro group. The catalyst can be readily recovered and reused for four cycles without significantly losing activity. The practicality of this study opens new avenues for heterogeneous Pd-catalyzed C—H functionalization chemistry.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was financially supported by the National Natural Science Foundation of China (No. 21861030) and the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (No. NJYT-17-A22).
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.02.055.
| [1] |
(a) N. Selander, K.J. Szabo', Chem. Rev. 111 (2011) 2048-2076; (b) V.G. Zaitsev, D. Shabashov, O. Daugulis, J. Am. Chem. Soc.127 (2005) 13154-13155; (c) A. Roglans, A. Pla-Quintana, M. Moreno-Maňas, Chem. Rev. 106 (2006) 4622-4643; (d) H.A. Chiong, Q.N. Pham, O. Daugulis, J. Am. Chem. Soc. 129 (2007) 9879-9884; (e) K.L. Hull, M.S. Sanford, J. Am. Chem. Soc. 131 (2009) 9651-9653; (f)D. Kalyani, N.R. Deprez, L.V. Desai, M.S. Sanford, J. Am. Chem. Soc.127 (2005) 7330-7331; (g) J. Park, M. Kim, S. Sharma, et al., Chem. Commun. 49 (2013) 1654-1656; (h)Y.Gao, Y.B.Huang, W.Q. Wu, K.Huang, H.F.Jiang, Chem. Commun. 50 (2014) 8370-8373. |
| [2] |
(a) Z. Huang, H.N. Lim, F. Mo, M.C. Young, G. Dong, Chem. Soc. Rev. 44 (2015) 7764-7786; (b) X. Chen, K.M. Engle, D.H. Wang, J.Q. Yu, Angew. Chem. Int. Ed. 48 (2009) 5094-5115; (c) D. Alberico, M.E. Scott, M. Lautens, Chem. Rev. 107 (2007) 174-238; (d) J.A. Labinger, J.E. Bercaw, Nature 417 (2002) 507-514; (e) B.D. Dangel, K. Godula, S.W. Youn, B. Sezen, D. Sames, J. Am. Chem. Soc. 124 (2002) 11856. |
| [3] |
(a) T.W. Lyons, M.S. Sanford, Chem. Rev. 110 (2010) 1147-1169; (b) O. Daugulis, J. Roane, L.D. Tran, Acc. Chem. Res. 48 (2015) 1053-1064; (c) G. He, B. Wang, W.A. Nack, G. Chen, Acc. Chem. Res. 49 (2016) 635-645; (d) O. Daugulis, H.Q. Do, D. Shabashov, Acc. Chem. Res. 42 (2009) 1074-1086; (e) O. Baudoin, Chem. Soc. Rev. 40 (2011) 4902-4911. |
| [4] |
(a) J.T. Joseph, J.C. Pablo, I.S. Noam, S.S. Melanie, Nature 531 (2016) 220-224; (b) G. Rouquet, N. Chatani, Angew. Chem. Int. Ed. 52 (2013) 11726-11743; (c) P.M. Vaibhav, J.A. Garcia-Lopez, ChemCatChem 9 (2017) 1149-1156; (d) B.J. Knight, J.O. Rothbaum, E.M. Ferreira, Chem. Sci. 7 (2016) 1982-1987; (e) Q. Zhang, B.F. Shi, Chin. J. Chem. 37 (2019) 647-656; (f) Z. Chen, B. Wang, J. Zhang, et al., Org. Chem. Front. 2 (2015) 1107-1295; (g) H. Li, B.J. Li, Z.J. Shi, Catal. Sci. Technol. 1 (2011) 191-206. |
| [5] |
(a) C.H. Jun, H. Lee, J.B. Hong, J. Org. Chem. 62 (1997) 1200-1201; (b) R.B. Bedford, S.J. Coles, M.B. Hursthouse, M.E. Limmert, Angew. Chem. Int. Ed. 42 (2003) 112-114; (c) F. Mo, G. Dong, Science 345 (2014) 68-72; (d) Q.J. Yao, S. Zhang, B.B. Zhan, B.F. Shi, Angew. Chem. Int. Ed. 56 (2017) 6617-6621; (e) G. Liao, Q.J. Yao, Z.Z. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 3661-3665; (f) G. Liao, B. Li, H.M. Chen, et al., Angew. Chem. Int. Ed. 57 (2018) 17151-17155; (g) G. Liao, H.M. Chen, Y.N. Xia, et al., Angew. Chem. Int. Ed. 58 (2019) 11464-11468; (h) S. Zhang, Q.J. Yao, G. Liao, et al., ACS Catal. 9 (2019) 1956-1961; (i) H.M. Chen, S. Zhang, G. Liao, et al., Organometallics 38 (2019) 4022-4028. |
| [6] |
F.L. Zhang, K. Hong, T.J. Li, H. Park, J.Q. Yu, Science 351 (2016) 252-256. DOI:10.1126/science.aad7893 |
| [7] |
F. Ma, M. Lei, L. Hu, Org. Lett. 18 (2016) 2708-2711. DOI:10.1021/acs.orglett.6b01170 |
| [8] |
K. Yang, Q. Li, Y. Liu, G. Li, H. Ge, J. Am. Chem. Soc. 138 (2016) 12775-12778. DOI:10.1021/jacs.6b08478 |
| [9] |
(a) D.T. D. Tang, K.D. Collins, F. Glorius, J. Am. Chem. Soc. 135 (2013) 7450-7453; (b) S. Squez-Cespedes, A. Ferry, L. Candish, F. Glorius, Angew. Chem. Int. Ed. 54 (2015) 5772-5776; (c) S. Korwar, M. Burkholder, S.E. Gilliland Ⅲ, et al., Chem. Commun. 53 (2017) 7022-7025; (d) Y.M. A. Yamada, Y. Yuyama, T. Sato, S. Fujikawa, Y. Uozumi, Angew. Chem. Int. Ed. 53 (2014) 127-131; (e) V.A. Zinovyeva, M.A. Vorotyntsev, I. Bezverkhyy, D. Chaumont, J.C. Hierso, Adv. Funct. Mater. 21 (2011) 1064-1075; |
| [10] |
(a) D. Zhang, B. Zhaorigetu, Y.S. Bao, J. Phys. Chem. C 119 (2015) 20426-20432; (b) Y.S. Bao, D. Zhang, M. Jia, B. Zhaorigetu, Green Chem. 18 (2016) 2072-2077. |
| [11] |
(a) V.G. Zaitsev, D. Shabashov, O. Daugulis, J. Am. Chem. Soc. 127 (2005) 13154-13155; (b) T. Neetipalli, D. Arnab, S. Anurag, et al., Adv. Synth. Catal. 361 (2019) 1441-1446. |
| [12] |
Y. Rozita, R. Brydson, T.P. Comyn, et al., ChemCatChem 5 (2013) 2695-2706. DOI:10.1002/cctc.201200880 |
| [13] |
T. Pillo, R. Zimmermann, P. Steiner, S. Hüfner, J. Phys. Condens. Matter 9 (1997) 3987-3999. |
| [14] |
K. Hong, H. Park, J.Q. Yu, ACS Catal. 7 (2017) 6938-6941. DOI:10.1021/acscatal.7b02905 |