 2021, Vol. 32
2021, Vol. 32 

b Department of Chemistry, University of Pennsylvania, PA 19104, United States;
c Department of Chemistry and Biochemistry, University of California, CA 90095, United States
Transition-metal-catalyzed cross-coupling reactions between organometallic reagents and organic halides, which have been widely applied due to their flexibility to construct new carbon-carbon bonds, comprise one of the most significant reaction types. Various organometallic cross-coupling partners (Negishi organozinc [1], Suzuki organoboron [2], Stille organotin [3], and Hiyama [4]/Denmark [5] organosilicon reagents), which in many cases are prepared from the corresponding organolithium species, have been well developed. In recent years, the Smith group reported a series of monomeric and polymer-supported siloxanes as effective and recyclable transfer agents (e.g., PSTA-I and PSTA-II, Fig. 1) that now permit the direct use of aryl and alkenyl organolithiums in the Pd-catalyzed cross-coupling reactions with aryl and alkenyl halides at room temperature [6]. Importantly, this type of cross-coupling reactions avoids the use of both stoichiometric base/fluorides and the formation of stoichiometric waste products. In addition, application of these siloxane transfer agents overcomes the intrinsic limitations of Murahashi [7] cross-coupling reactions and the need for the Feringa [8] protocol, in which organolithium reagents require slow addition to prevent competing formation of homo-coupled products caused by rapid lithium-halogen exchange.

|
Download:
|
| Fig. 1. Cross-coupling reactions of organolithium reagents mediated by siloxane transfer agents. | |
{kind=link}
As shown in Fig. 1, in this Pd-catalyzed cross-coupling reaction, a catalytic amount of CuI is required. Previous studies by the Hiyama [9] and Tamao [10] groups also found that copper(Ⅰ) salts play an important role in similar systems. They proposed that these palladium- and copper-catalyzed reactions involve two transmetalation steps: the first transmetalation from silicon to copper(Ⅰ) and the second transmetalation from copper(Ⅰ) to palladium(Ⅱ). However, it was not clear what copper species is involved in the transmetalation, nor has a detailed mechanism of this siloxane-mediated Pd-catalyzed cross-coupling reaction been elucidated.
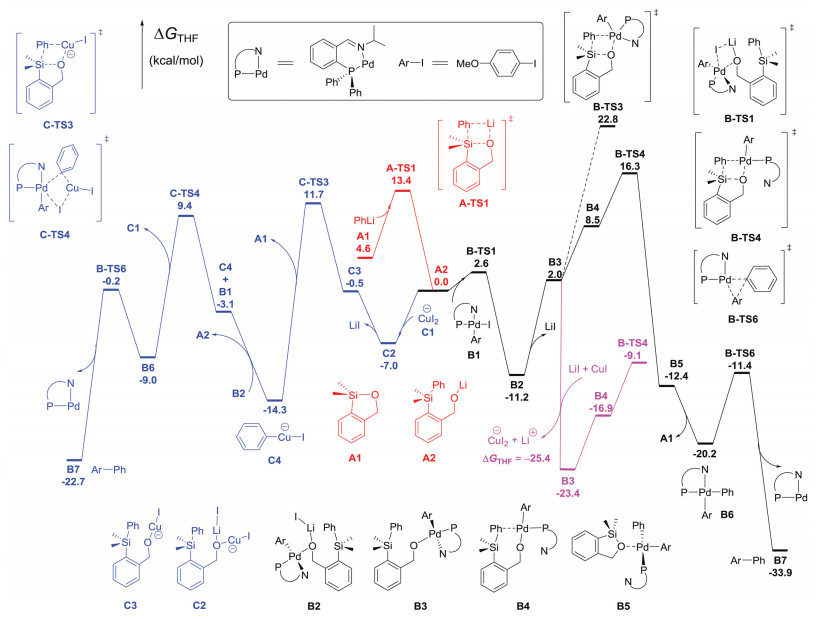
We explored computationally the mechanism of the Pd-catalyzed cross-coupling reaction of organolithium reagents and aryl iodides mediated by a siloxane transfer agent, and in particular, the role of CuI during the transmetalation processes, using density functional theory (DFT) calculations. Geometries of minima and transition-state structures are optimized at the B3LYP/6-31G(d)[SDD, for Cu, Pd, and I] level. Solvent effects in THF were evaluated on the gas-phase optimized structures using the PCM model at the M06/6-311+G(d, p)[SDD, for Cu, Pd, and I] level. The results are summarized in Fig. 2, with the key structures illustrated in Fig. 3.

|
Download:
|
| Fig. 2. DFT-computed free energies for the siloxane-mediated Pd-catalyzed cross-coupling reaction of para-methoxy-iodobenzene with phenyllithium in the absence or presence of CuI. Reactions begin at transfer agent A1 in the center of the diagram. | |
{kind=link}

|
Download:
|
| Fig. 3. DFT-optimized key structures for the siloxane-mediated Pd-catalyzed cross-coupling reaction of para-methoxy-iodobenzene with phenyllithium in the absence or presence of CuI (distances are given in Å). | |
{kind=link}
Fig. 2 (red lines) shows that nucleophilic attack of phenyllithium on the siloxane transfer agent A1 at the silicon center via transition state A-TS1 requires an activation free energy of 8.8 kcal/mol in THF. The formation of lithium alkoxide A2 is exergonic by 4.6 kcal/mol in THF. This indicates that by using siloxanes as transfer agents, highly reactive organolithium reagents can be efficiently converted into relatively stable organosilicon species. The stability of A2 overcomes the problems caused by fast lithium-halogen exchange, and avoids undesired homo-coupled products.
The black lines in Fig. 2 depict the DFT-computed free energies for the Pd-catalyzed cross-coupling reaction of para-methoxy-iodobenzene with organosilicon intermediate A2 in the absence of CuI. Previous studies demonstrated that the oxidative addition of aryl iodides to Pd(0) catalysts is very facile [11]. The generated Ar[Pd(Ⅱ)]I species B1 can, in turn, react with lithium alkoxide A2 to form a complex B2 via transition state B-TS1, which requires an activation free energy of only 2.6 kcal/mol. As shown in Fig. 3, in the transition-state structure B-TS1, the forming O-Pd and breaking I-Pd distances are 2.90 and 2.85 Å, respectively, and the O-Pd-I angle is 84°. This transition state could be viewed as an organometallic analog of the frontside SN2 substitution [12] in organic chemistry. The formation of complex B2 is exergonic by 11.2 kcal/mol in THF, indicating that there is no free Ar[Pd(Ⅱ)]I species B1 in the system. From B2, the Si-Pd(Ⅱ) transmetalation is a three-step process. The first step is the dissociation of LiI from complex B2. As shown in Fig. 3, the LiI moiety in B2 is not only coordinated by oxygen, but also located between the p-MeOPh group and the Ph group with the shortest C-Li distances of 2.99 and 2.61 Å, respectively. Previous studies indicated that there is significant cation-π interaction between Li+ and arenes [13]. Therefore, the dissociation of LiI from complex B2 to generate intermediate B3 is endergonic by 13.2 kcal/mol in THF, which is quite unfavorable thermodynamically. The second step is the replacement of nitrogen coordination at the palladium center by the phenyl group attached to the silicon atom. Since the arene in B4 (Fig. 3, C-Pd distance of 2.63 Å) is a weak ligand to palladium as compared to the imine in B3, this ligand exchange step is also unfavorable, increasing the reaction free energy by 6.5 kcal/mol. The third step is the phenyl group transfer from silicon to palladium. This step requires an activation free energy of 7.8 kcal/mol via the σ-bond metathesis transition state B-TS4. Fig. 3 shows the geometries of the Pd-O-Si-C(Ph) four-membered ring in B4 and B-TS4. From B4 to B-TS4, the C(Ph)-Si distance increases from 1.93 Å to 2.15 Å, while the C(Ph)-Pd distance decreases from 2.63 Å to 2.29 Å. Additionally, the Si-O distance decreases from 2.37 Å to 1.83 Å, and the Pd-O distance slightly increases from 2.12 Å to 2.20 Å. From complex B2 (Fig. 2, -11.2 kcal/mol) to transition state B-TS4 (16.3 kcal/mol), the overall barrier for the three-step Si-Pd(Ⅱ) transmetalation is 27.5 kcal/mol, with the generation of complex B5 (-12.4 kcal/mol) exergonic by 1.2 kcal/mol in THF. The frontside SN2 substitution transmetalation transition state B-TS3 (Fig. 2, 22.8 kcal/mol) involving five-coordinated palladium center is also located, which is 6.5 kcal/mol higher than the σ-bond metathesis transition state B-TS4 (16.3 kcal/mol) involving four-coordinated palladium center. The decomposition of complex B5 leads to the regeneration of siloxane transfer agent A1 and the formation of Ar[Pd(Ⅱ)]Ph species B6, which is exergonic by 7.8 kcal/mol. The final step entails the Ar[Pd(Ⅱ)]Ph species B6 undergoing reductive elimination to regenerate the Pd(0) catalyst and to form the cross-coupling product B7 via transition state B-TS6. This step has a low activation free energy of 8.8 kcal/mol and is exergonic by 13.7 kcal/mol in THF. Overall, the rate-determining step for this Pd-catalyzed cross-coupling reaction (without CuI) is the Si-Pd(Ⅱ) transmetalation from B2 to B-TS4, requiring a total activation free energy of 27.5 kcal/mol. Such a barrier is too high for the reaction to occur at room temperature. Moreover, the overall barrier for the regeneration of PhLi (from B2 to A-TS1, Fig. 2) is 24.6 kcal/mol, which is 2.9 kcal/mol lower than that for the cross-coupling reaction (27.5 kcal/mol). This may result in competing formation of the undesired homo-coupled products.
As previously mentioned, the dissociation of LiI during the Si-Pd(Ⅱ) transmetalation is thermodynamically disfavored, contributing 13.2 kcal/mol in overall barrier (27.5 kcal/mol). By contrast, calculations indicate that the reaction of LiI with CuI to form Li+ and CuI2- is highly exergonic by 25.4 kcal/mol in THF. Therefore, when a catalytic amount of CuI is present in the reaction system, the initial role of CuI is to react with the dissociated LiI, resulting in the formation of intermediate B3 exergonic by 12.2 kcal/mol (Fig. 2, pink lines). The subsequent steps from B3 to the σ-bond metathesis transition state B-TS4 require an overall activation free energy of 14.3 kcal/mol. This dramatically accelerates the Si-Pd(Ⅱ) transmetalation, making the reaction smoothly occur at room temperature. When the catalytic amount of CuI (usually 10 mol%) is completely converted into CuI2-, such a reaction pathway does not work at all.
The blue lines in Fig. 2 show the DFT-computed free energies for the CuI2- and Pd-cocatalyzed cross-coupling reaction. The organosilicon intermediate A2 reacts with CuI2- to form complex C2, which is exergonic by 7.0 kcal/mol in THF. The Si-Cu(Ⅰ) transmetalation starts from the dissociation of LiI in C2 to form intermediate C3, which then undergoes the phenyl group transfer from silicon to palladium via the σ-bond metathesis transition state C-TS3 (Fig. 3, C-Si and C-Cu distances of 2.35 Å and 2.04 Å, respectively). The overall barrier for the Si-Cu(Ⅰ) transmetalation is 18.7 kcal/mol, with the regeneration of siloxane transfer agent A1 and the formation of PhCuI- (C4) exergonic by 7.3 kcal/mol. The free Ar[Pd(Ⅱ)]I species B1 can be generated from B2 with the increase of reaction free energy by 11.2 kcal/mol. Then, the Cu(Ⅰ)-Pd(Ⅱ) transmetalation [14] between PhCuI- and Ar[Pd(Ⅱ)]I occurs via the frontside SN2 substitution transition state C-TS4 (Fig. 3, C-Cu and C-Pd distances of 1.95 and 3.45 Å, respectively; a Pd-I distance of 2.89 Å; and a C-Pd-I angle of 92°). The overall barrier for the Cu(Ⅰ)-Pd(Ⅱ) transmetalation is 23.7 kcal/mol, leading to the regeneration of the CuI2- catalyst and the formation of Ar[Pd(Ⅱ)]Ph species B6, which can easily undergo reductive elimination to give the final product Ar-Ph (B7) and to regenerate the Pd(0) catalyst. Therefore, the rate-determining step for this CuI2- and Pd-cocatalyzed cross-coupling reaction is the Cu(Ⅰ)-Pd(Ⅱ) transmetalation from C4 to C-TS4 with a total activation free energy of 23.7 kcal/mol in THF. This barrier is about 4 kcal/mol lower than that of the solely Pd-catalyzed reaction (without CuI, 27.5 kcal/mol). Notably, in this reaction system, the regeneration of PhLi from C4 to A-TS1 (Fig. 2) requires an overall activation free energy of 27.7 kcal/mol, which is about 4 kcal/mol higher than that of the cross-coupling reaction (23.7 kcal/mol). This effectively inhibits the formation of the undesired homo-coupled products.
In conclusion, the detailed mechanisms of the Pd-catalyzed cross-coupling reaction of aryl lithium mediated by the siloxane transfer agent in the absence or presence of CuI have been investigated by DFT calculations. We find that a catalytic amount of CuI first accelerates the Si-Pd(Ⅱ) transmetalation by the highly exergonic reaction with LiI to form CuI2-. Subsequently, CuI2- works as a shuttle between the Si-Cu(Ⅰ) and Cu(Ⅰ)-Pd(Ⅱ) transmetalation processes [15], decreasing the overall barrier for the cross-coupling reaction significantly. This theoretical finding may be applicable to other CuI-promoted cross-coupling reactions.
Declaration of competing interestNo competing financial interest.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 21803030), the Fundamental Research Funds for the Central Universities, the National Thousand Young Talents Program, the Jiangsu Innovation & Entrepreneurship Talents Plan, and the NSF of Jiangsu Province (No. BK20170631) in China, the National Institutes of Health (No. CA-019033) and the National Science Foundation in USA (No. CHE-1361104).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.05.009.
| [1] |
(a) E. Negishi, S. Baba, Chem. Commun. (Camb. ) (1976) 596-597; (b) S. Baba, E. Negishi, J. Am. Chem. Soc. 98 (1976) 6729-6731; (c) E. Negishi, Acc. Chem. Res. 15 (1982) 340-348. |
| [2] |
(a) N. Miyaura, K. Yamada, A. Suzuki, Tetrahedron Lett. 20 (1979) 3437-3440; (b) N. Miyaura, A. Suzuki, Chem. Commun. (Camb. ) (1979) 866-867; (c) N. Miyaura, A. Suzuki, Chem. Rev. 95 (1995) 2457-2483. |
| [3] |
(a) D. Milstein, J.K. Stille, J. Am. Chem. Soc. 100 (1978) 3636-3638; (b) D. Milstein, J.K. Stille, J. Am. Chem. Soc. 101 (1979) 4992-4998; (c) J.K. Stille, Angew. Chem. Int. Ed. Engl. 25 (1986) 508-524. |
| [4] |
(a) Y. Hatanaka, T. Hiyama, Synlett (1991) 845-853; (b) T. Hiyama, T. Hatanaka, Pure Appl. Chem. 66 (1994) 1471-1478; (c) J. Chen, M. Tanaka, A.K. Sahoo, et al., Bull. Chem. Soc. Jpn. 83 (2010) 554-569; (d)Y. Nakao, M. Takeda, T. Matsumoto, T. Hiyama, Angew. Chem. Int. Ed. 49 (2010) 4447-4450. |
| [5] |
(a) S.E. Denmark, J.Y. Choi, J. Am. Chem. Soc. 121 (1999) 5821-5822; (b) S.E. Denmark, R.F. Sweis, J. Am. Chem. Soc. 126 (2004) 4876-4882; (c) S.E. Denmark, R.F. Sweis, Acc. Chem. Res. 35 (2002) 835-846; (d) S.E. Denmark, C.S. Regens, Acc. Chem. Res. 41 (2008) 1486-1499. |
| [6] |
(a) A.B. Smith Ⅲ, A.T. Hoye, D. Martinez-Solorio, W.S. Kim, R. Tong, J. Am. Chem. Soc. 134 (2012) 4533-4536; (b) D. Martinez-Solorio, A.T. Hoye, M.H. Nguyen, A.B. Smith Ⅲ, Org. Lett. 15 (2013) 2454-2457; (c) M.H. Nguyen, A.B. Smith Ⅲ, Org. Lett. 15 (2013) 4258-4261; (d) M.H. Nguyen, A.B. Smith Ⅲ, Org. Lett. 16 (2014) 2070-2073; (e) D. Martinez-Solorio, B. Melillo, L. Sanchez, et al., J. Am. Chem. Soc. 138 (2016) 1836-1839; (f) M.H. Nguyen, K.T. O'Brien, A.B. Smith Ⅲ, J. Org. Chem. 82 (2017) 11056-11071. |
| [7] |
(a) S.I. Murahashi, Y. Tanba, M. Yamamura, I. Moritani, Tetrahedron Lett. 15 (1974) 3749-3752; (b) S.I. Murahashi, Y. Tanba, M. Yamamura, N. Yoshimura, J. Org. Chem. 43 (1978) 4099-4106; (c) S.I. Murahashi, J. Organomet. Chem. 653 (2002) 27-33. |
| [8] |
M. Giannerini, M. Fananas-Mastral, B.L. Feringa, Nat. Chem. 5 (2013) 667-672. DOI:10.1038/nchem.1678 |
| [9] |
Y. Nakao, T. Hiyama, Chem. Soc. Rev. 40 (2011) 4893-4901. DOI:10.1039/c1cs15122c |
| [10] |
E.C. Son, H. Tsuji, T. Saeki, K. Tamao, Bull. Chem. Soc. Jpn. 79 (2006) 492-494. DOI:10.1246/bcsj.79.492 |
| [11] |
K.J. Bonney, F. Schoenebeck, Chem. Soc. Rev. 43 (2014) 6609-6638. DOI:10.1039/C4CS00061G |
| [12] |
(a) F.M. Bickelhaupt, J. Comput. Chem. 20 (1999) 114-128; (b) A.P. Bento, F.M. Bickelhaupt, J. Org. Chem. 73 (2008) 7290-7299; (c) A.P. Bento, F.M. Bickelhaupt, Chem. Asian J. 3 (2008) 1783-1792. |
| [13] |
(a) J.B. Nicholas, B.P. Hay, D.A. Dixon, J. Phys. Chem. A 103 (1999) 1394-1400; (b) D. Feller, D.A. Dixon, J.B. Nicholas, J. Phys. Chem. A 104 (2000) 11414-11419; (c) J.C. Amicangelo, P.B. Armentrout, J. Phys. Chem. A 104 (2000) 11420-11432; (d) S. Tsuzuki, M. Yoshida, T. Uchimaru, M. Mikami, J. Phys. Chem. A 105 (2001) 769-773; (e) D. Kim, S. Hu, P. Tarakeshwar, K.S. Kim, J. Phys. Chem. A107 (2003)1228-1238; (f) D.A. Dougherty, Acc. Chem. Res. 46 (2013) 885-893. |
| [14] |
H. Zheng, K. Semba, Y. Nakao, S. Sakaki, J. Am. Chem. Soc. 139 (2017) 14065-14076. DOI:10.1021/jacs.7b04383 |
| [15] |
J. dePozo, D. Carrasco, M.H. Pérez-Temprano, et al., Angew. Chem. Int. Ed. 52 (2013) 2189-2193. DOI:10.1002/anie.201209262 |