 2021, Vol. 32
2021, Vol. 32 


b State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences of Guangxi Normal University, Guilin 541004, China
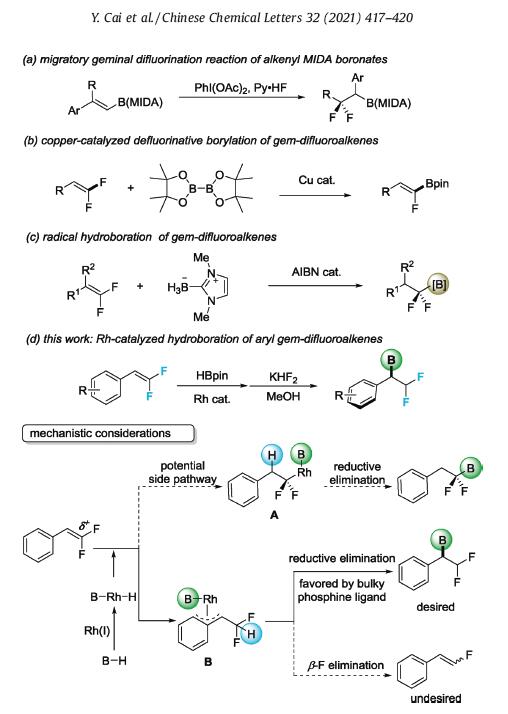
Organofluorines have found wide spread applications in pharmaceuticals, agrochemicals, and advanced functional materials [1]. In particular, the difluoromethyl group, as a chemically stable bioisostere of hydroxy and mercapto in medicinal chemistry, has gained substantial attention [2]. Historically, difluoromethyl group could be synthesized via the deoxyfluorination of aldehydes with dialkylaminosulfur trifluorides [3]. The drastic reaction conditions restrict the functional group compatibility of this method. Recently, more efforts have been paid on the use of difluoromethylene-containing building blocks, as represented by (pseudo)-halo difluoromethane [4], difluoromethyl metallic [5], difluoromethyl sulfinate [6] and other masked CHF2 equivalent [7], for the modular difluoromethylation reaction. On the other hand, the direct difluorination of alkenes promoted by hypervalent iodine provides another useful approach for difluoromethylated compounds synthesis [8]. Inspired by this strategy, we recently realized an efficient synthesis of -difluoromethylated alkyl boron starting from aryl-substituted alkenyl N-methyliminodiacetyl (MIDA) boronates (Scheme 1a) [9]. The boron moiety is retained to the product and thus brings about additional value for further derivatizations. In light of the utility of borylated organofluorines in organic synthesis, we [10] and others [11] independently reported the copper-catalyzed defluorinative borylation of gem-difluoroalkenes with B2pin2, leading to monofluoroalkenyl boron compounds (Scheme 1b). A facile copper-mediated -F elimination occurs in these processes. Further, the radical boryl addition to gem-difluoroalkenes delivers another type of -difluorinated alkylboron compounds, as recently disclosed by Wang [12a] and us [12b] (Scheme 1c).

|
Download:
|
| Scheme 1. Representative methods to access fluorinated organoborons. | |
Rh-catalyzed hydroboration of alkenes has attracted great attention as a powerful synthetic approach to organoboron compounds [13]. In connection with our previous studies on the synthesis of borylated organofluorines, we proposed the hydroboration of gem-difluoroalkenes might provide a chance to deliver the difluoromethylated benzylborons (Scheme 1d). On one hand, we reasoned the biased electron-distribution caused by the fluorine effect should render a high regioselectivity. That is, the hydride in Rh(III)-H bond, formed by oxdative B—H bond addition, would be preferentially attached to the electron-deficient α-C in the migratory insertion step. On the other hand, the formation of stable η-3-benzyl metal complex B when aryl gem-difluoroalkenes are used could be another important factor for high regioselectivity [14], especially when cationic rhodium catalyst were used [13g]. Nevertheless, challenges remain given that the metal-mediated β-F elimination was proposed to be facile in many reactions. Thus, the C—B reductive elimination from B should outpace the β-F elimination step for effective reaction. Herein, we report that by fine tuning the phosphine ligand, a cationic rhodium(I)-catalyzed hydroboration of gem-difluoroalkenes was realizable to provide a diverse array of difluoromethylated benzylborons.
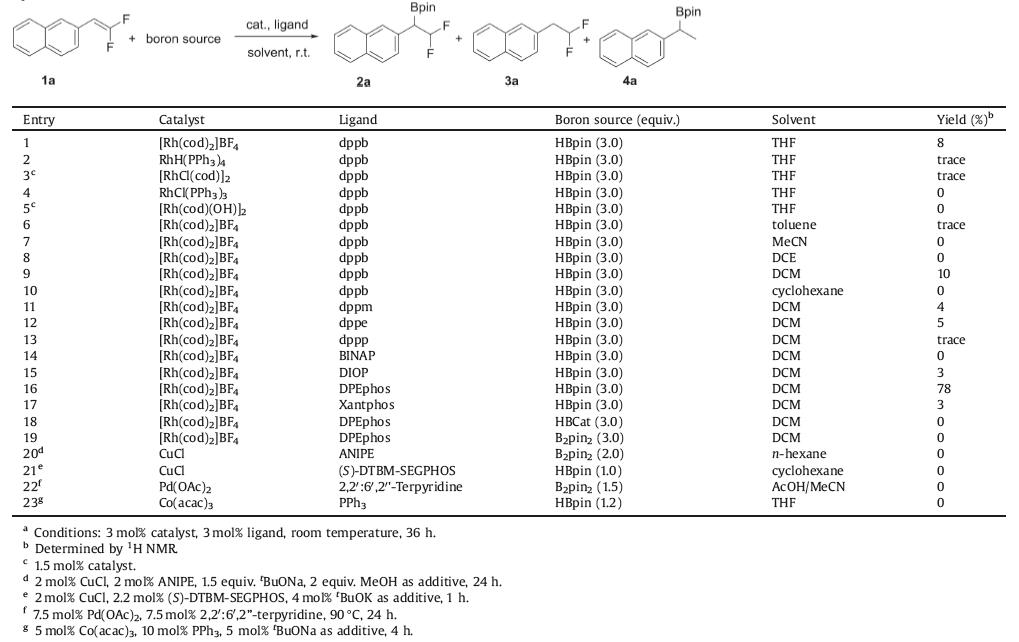
The hydroboration reaction was carried out by reacting 2-(2, 2-difluorovinyl)naphthalene 1a with pinacol borane (H-Bpin) as the hydroborating reagent (Table 1). It was found the use of cationic [Rh(cod)2]BF4 as catalyst and dppb (for structure of ligand, Fig. 1) as ligand in THF, the reaction indeed delivered the desired hydroboration product 2a, although in a low 8% yield (entry 1). The double bond hydrogenation (3a) and defluorinative hydroboration (4a) products were determined to be major side products. While rhodium-based catalysts including RhH(PPh3)4, [Rh(cod)Cl]2 showed trace reactivity, catalysts RhCl(PPh3)3 and [Rh(cod)(OH)]2 resulted in an inseparable mixture, with no desired product being formed (entries 2—5). Solvent screening revealed DCM is optimal, giving a slightly better yield of 10% (entries 6—10). The ligand effect in this protocol is pronounced. Biphosphine ligands with small bite angle were ineffective in this reaction, leading predominately to the recovery of the starting material (entries 11—15). We found that the use of DPEphos with larger bite angle improved the yield dramatically to 78% (entry 16), probably by facilitate the desirable reductive C—B bond elimination as shown in Scheme 1d. However, further increase the bite angle of the ligand was proven unfruitful, as the use of Xantphos led to a decreased yield of 3% (entry 17). The displacement of H-Bpin with other boron sources such as HBCat (entry 18) and B2pin2 (entry 19) failed to give any hydroboration product. The use of other metal based catalytic systems, including copper [15], palladium [16], and cobalt [17] which have been previously demonstrated to be efficient in alkene hydroboration reaction, all failed to give satisfactoryresults. The product was sensitive to silica gel column chromatography since a facile elimination of B—F bond occurs to deliver a monofluoroalkene side product. Thus, the conversion of the product to its potassium trifluorobronates congener was conducted prior to the isolation of chemically pure product via flash chromatography. Some chiral phosphine ligand, such as (S)-DIOP, (S)-DTBM-SEGPHOS and (R)-BINAP were also test. Unfortunately, trivial enantioselectivity was found.
|
|
Table 1 Optimization of the reaction.a |
{kind=link}

|
Download:
|
| Fig. 1. Ligands used in this study. | |
{kind=link}
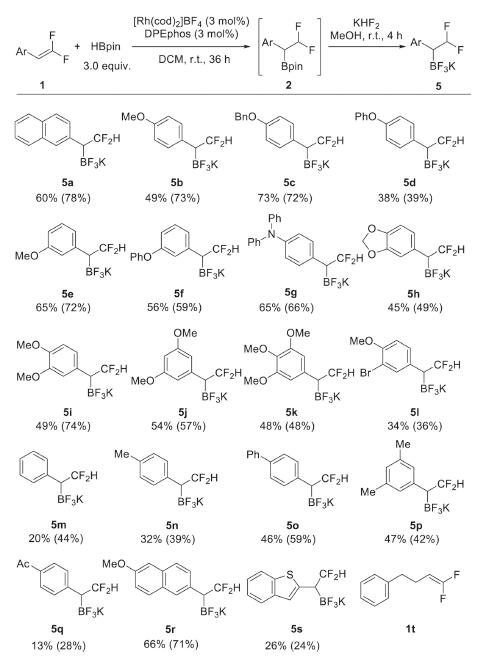
With the optimized reaction conditions in hand (Table 1, entry 16), we set out to investigated the generality and limitation of the reaction. As shown in Scheme 2, aryl gem-difluoroalkenes bearing electron-donating oxygen- (5b-5f) or nitrogen- (5g) centered substituent produced the corresponding products in moderate to good yields. The multi alkoxyl-substituted substrates were also compatible well (5h-5k). Comparatively, the electron-neutral aryl substituent led to lower yields (5m-5p). When electron-poor substrates were used, a significant amount of hydrogenation product derived from the protonation of the benzylic rhodium intermediate was observed. As such, the acetyl-substituted substrate delivered the desired product 5q in only 13% isolated yield. As expected, the methoxy-substituted naphthalene substrate gave a good yield of 66% (5r). A benzo[b]thiophenyl group was also tolerated, although in low yield (5s). We have also tried acetyl-protected indole derivative for this reaction, however, no desired product was formed. The alkyl-substituted gem-difluoroalkene 1t was unfortunately not applicable to the reaction.

|
Download:
|
| Scheme 2. Substrate scope. Reaction conditions: 1 (0.2 mmol, 1.0 equiv.), HBpin (0.6 mmol), [Rh(cod)2]BF4 (3 mol%), DPEphos (3 mol%), DCM (0.6 mL) at room temperature under an argon atmosphere for 36 h. Then, KHF2, MeOH, r.t., 4 h. Isolated yield of 5 over two steps. Yields in brackets are yields of 2, determined by 1H NMR spectroscopy using 1-iodo-4-methoxybenzene as an internal standard. | |
{kind=link}
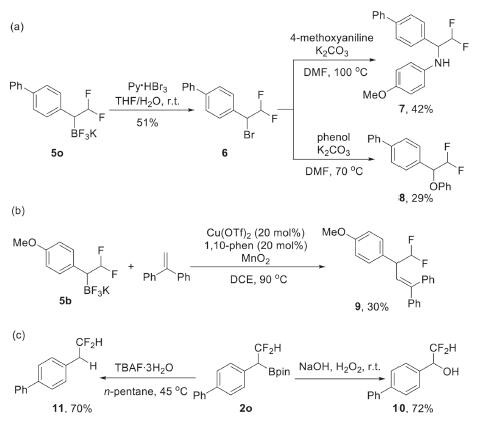
Efforts were then paid to the derivatization of the products. The oxidation of C—B in 5o with Py·HBr3 led to a difluoromethylated benzyl bromide 6, which could be further transformed to α-difluoromethylated amine 7 or ether 8 or via nucleophilic substitution reaction (Scheme 3a). A copper-catalyzed oxidative radical coupling of C—B bond with 1, 2-diphenylethylene was also feasible to furnish a new C—C bond (Scheme 3b). Starting from pinacolboronate 2o, the oxidation with H2O2 provided a benzyl alcohol 10 in 72% yield, and the treatment with TBAF resulted in a protodeborylation product 11 (Scheme 3c).

|
Download:
|
| Scheme 3. Derivatization of the products. | |
{kind=link}
In conclusion, a catalytic hydroboration of aryl gem-difluoroalkenes with a cationic rhodium catalyst and a large bite angle biphosphine ligand was developed. The catalytic system offers good regioselectivity and diminishes the undesired β-F elimination to a large extent. A number of electron-rich aryl gem-difluoroalkenes were smoothly converted to the α-difluoromethylated benzyl borons in reasonable yields. A modular synthesis of diverse difluoromethylated compounds were demonstrated from the product obtained via this protocol.
Declaration of competing interestThe authors declare no competing financial interest.
AcknowledgmentsGenerous financial support from the National Natural Science Foundation of China (No. 21971261), Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery (No. 2019B030301005), Key Project of Chinese National Programs for Fundamental Research and Development (No. 2016YFA0602900), and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (No. 2017BT01Y093) is gratefully acknowledged.
| [1] |
(a) K. Muller, C. Faeh, F. Diederich, Science 317 (2007) 1881-1886; (b) S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320-330; (c) W.K. Hagmann, J. Med. Chem. 51 (2008) 4359-4369; (d) J. Wang, M. Sanchez-Rosello, J.L. Acena, et al., Chem. Rev. 114 (2014) 2432-2506. |
| [2] |
(a) J.A. Erickson, J.I. McLoughlin, J. Org. Chem. 60 (1995) 1626-1631; (b) N.A. Meanwell, J. Med. Chem. 54 (2011) 2529-2591; (c) J. Rong, C. Ni, J. Hu, Asian J. Org. Chem. 6 (2017) 139-152; (d) Z. Feng, Y.L. Xiao, X. Zhang, Acc. Chem. Res. 51 (2018) 2264-2278. |
| [3] |
(a) W.J. Middleton, J. Org. Chem. 40 (1975) 574-578; (b) J.M. Shreeve, R.P. Singh, Synthesis (2002) 2561-2578; (c) P.R. Melvin, D.M. Ferguson, S.D. Schimler, D.C. Bland, M.S. Sanford, Org. Lett. 21 (2019) 1350-1353. |
| [4] |
(a) Z. Feng, Q.Q. Min, X.P. Fu, L. An, X. Zhang, Nat. Commun. 9 (2017) 918-923; (b) C. Xu, W.H. Guo, X. He, et al., Nat. Chem. 9 (2018) 1170; (c) W. Miao, Y. Zhao, C. Ni, et al., J. Am. Chem. Soc. 140 (2018) 880-883. |
| [5] |
(a) X. Zeng, W. Yan, S.B. Zacate, et al., J. Am. Chem. Soc. 141 (2019) 11398-11403; (b) Y. Zhao, W. Huang, J. Zheng, J. Hu, Org. Lett. 13 (2011) 5342-5345; (c) P.S. Fier, J.F. Hartwig, J. Am. Chem. Soc. 134 (2012) 5524-5527; (d) G.K. Prakash, S.K. Ganesh, J.P. Jones, et al., Angew. Chem. Int. Ed. 51 (2012) 12090-12094; (e) J.R. Bour, S.K. Kariofillis, M.S. Sanford, Organometallics 36 (2017) 1220-1223. |
| [6] |
Y. Fujiwara, J.A. Dixon, F. O'Hara, et al., Nature 492 (2012) 95-99. DOI:10.1038/nature11680 |
| [7] |
(a) T. Yokomatsu, T. Murano, K. Suemune, S. Shibuya, Tetrahedron 53 (1997) 815-822; (b) Q. Xie, Z. Zhu, L. Li, C. Ni, J. Hu, Angew. Chem. Int. Ed. 58 (2019) 6405-6410; (c) X. Peng, M.Y. Xiao, J.L. Zeng, F.G. Zhang, J. A. Ma, Org. Lett. 21 (2019) 4808-4811. |
| [8] |
(a) S.V. Kohlhepp, T. Gulder, Chem. Soc. Rev. 45 (2016) 6270-6288; (b) N.O. Ilchenko, B.O. Tasch, K.J. Szabo, Angew. Chem. Int. Ed. 53 (2014) 12897-12901; (c) F. Scheidt, J. Neufeld, M. Schafer, C. Thiehoff, R. Gilmour, Org. Lett. 20 (2018) 8073-8076; (d)T. Kitamura, K. Muta, J. Oyamada, J. Oyamada, J. Org. Chem. 80 (2015) 10431-10436; (e) S.M. Banik, J.W. Medley, E.N. Jacobsen, Science 353 (2016) 51-54; (f) Z. Zhao, L. Racicot, G.K. Murphy, Angew. Chem. Int. Ed. 56 (2017) 11620-11623. |
| [9] |
W.X. Lv, Q. Li, J.L. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 16544-16548. DOI:10.1002/anie.201810204 |
| [10] |
D.H. Tan, E. Lin, W.W. Ji, et al., Adv. Synth. Catal. 360 (2018) 1032-1037. DOI:10.1002/adsc.201701497 |
| [11] |
(a) J. Zhang, W. Dai, Q. Liu, S. Cao, Org. Lett. 19 (2017) 3283-3286; (b) H. Sakaguchi, Y. Uetake, M. Ohashi, et al., J. Am. Chem. Soc. 139 (2017) 12855-12862; (c) H. Sakaguchi, M. Ohashi, S. Ogoshi, Angew. Chem. Int. Ed. 57 (2018) 328-332; (d) J. Hu, X. Han, Y. Yuan, Z. Shi, Angew. Chem. Int. Ed. 56 (2017) 13342-13346; (e) R. Kojima, K. Kubota, H. Ito, Chem. Commun. 53 (2017) 10688-10691. |
| [12] |
(a) J.K. Jin, W.X. Zheng, H.M. Xia, F.L. Zhang, Y.F. Wang, Org. Lett. 21 (2019) 8414-8418; (b) X. Liu, E.E. Lin, G. Chen, et al., Org. Lett. 21 (2019) 8454-8458. |
| [13] |
(a) D. Männig, H. Nöth, Angew. Chem. Int. Ed. 24 (1985) 878-879; (b) D.A. Evans, G.C. Fu, A.H. Hoveyda, J. Am. Chem. Soc. 110 (1988) 6917-6918; (c) T. Hayashi, Y. Matsumoto, Y. Ito, J. Am. Chem. Soc. 111 (1989) 3426-3428; (d) D.R. Edwards, Y.B. Hleba, C.J. Lata, L.A. Calhoun, C.M. Crudden, Angew. Chem. Int. Ed. 46 (2007) 7799-7802; (e) J.R. Smith, B.S.L. Collins, M.J. Hesse, et al., J. Am. Chem. Soc. 139 (2017) 9148-9151; (f) G.L. Hoang, J.M. Takacs, Chem. Sci. 8 (2017) 4511-4516; (g) C.M. Crudden, Y.B. Hleba, A.C. Chen, J. Am. Chem. Soc. 126 (2004) 9200-9201; (h) Y. Hu, W. Sun, C. Liu, Synlett 30 (2019) 1105-1110. |
| [14] |
B.M. Trost, L.C. Czabaniuk, Angew. Chem. Int. Ed. 53 (2014) 2826-2851. DOI:10.1002/anie.201305972 |
| [15] |
(a) Y. Xi, J.F. Hartwig, J. Am. Chem. Soc. 139 (2017) 12758-12772; (b) Y. Cai, X.T. Yang, S.Q. Zhang, et al., Angew. Chem. Int. Ed. 57 (2018) 1376-1380. |
| [16] |
J. Huang, W. Yan, C. Tan, W. Wu, H. Jiang, Chem. Commun. 54 (2018) 1770-1773. DOI:10.1039/C7CC09432A |
| [17] |
(a) S.R. Tamang, D. Bedi, S. Shafiei-Haghighi, et al., Org. Lett. 20 (2018) 6695-6700; (b) J. Chen, J. Guo, Z. Lu, Chin. J. Chem. 36 (2018) 1075-1109. |