 2021, Vol. 32
2021, Vol. 32 


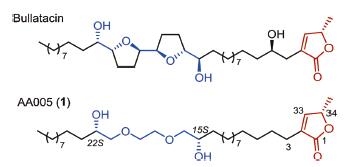
Annonaceous acetogenins (ACGs) are a large family of naturally occurring polyketides displaying extraordinary potent cytotoxicities [1]. However, low natural abundance, presence of multiple stereocenters, and poorly selective higher cytotoxicities (e.g., natural bullatacin (Fig. 1) against KB cell line with an IC50 < 10-12 μg/mL) [2], prevent them from further biological and pharmaceutical development. In the past two decades, a number of structural modifications and mimicries of natural ACGs have been studied to improve the action-selectivity of natural products, and simplify the complex natural structure as well [3, 4]. Among these efforts, AA005 (1, Fig. 1), a simplified analogue of natural bullatacin, is one outstanding representative developed by our group through continuous biological screenings and optimizations [4]. AA005 (1) not only exerts comparable activity as that of its natural parent, but also shows pronounced action-selectivity against the proliferation of cancer cells by comparison with corresponding normal cells [4c]. More recently, our study revealed that AA005 (1) shows imposing antitumor activity in vitro and in vivo by induction of cell death, which occurs in association with downregulation of Mcl-1 and the AIF-dependent signaling pathway [4g], showing the potential for further development of new pharmaceuticals. To meet the needs of various animal experiments, large-quantity supply of AA005 (1) is of urgency. Unfortunately, the previous synthesis is only capable to support cell-based screening and cell studies, affording relatively low quantity of the material [4a]. Herein, we wish to report a new ten-step scalable laboratory synthesis of AA005 (1) commencing with economically available materials ethylene glycol, (R)-epichlorohydrin, ethyl L-lactate and undecenoic acid.

|
Download:
|
| Fig. 1. Naturally occurring annonaceous acetogenin bullatacin and mimicking compound AA005 (1). | |
{kind=link}
AA005 (1, Fig. 1) retains the terminal γ-methylated butenolide, a common functionality of most natural ACGs, and replaces the middle bis-THF core with a simpler triglycol ether moiety. Such a modification greatly simplifies the structural complexity, as well as its chemical synthesis. Two strategies have been developed and extensively applied into the preparation of the γ-methylated butenolide of either natural ACGs or unnatural mimics [5], including the one disclosed by our laboratory [5b,5c]. Different chiral pools have been utilized for the α-alkylation of corresponding carbonyl compounds in the two approaches, resulting in diverse protocols to generate the requisite conjugate C=C bond. Of note, racemic phenomena of different degrees often happens under basic conditions at the stereogenic allylic C34 [6], which is usually introduced from proper chiral materials. Avoiding or minimizing use of basic treatments is preferred after generating this unsaturated-lactone moiety in the synthesis. The other characteristic feature of AA005 (1) is its middle subunit of C2-symmetrical triglycol flanking with two chiral secondary alcohols. Since the first report in 2000 [4a], a number of optimizations have been made for the synthesis of this triglycol unit of AA005 (1) [4b,4d,4f]. In a recent work, both the two chiral hydroxyls of AA005 (1) were introduced by a chiron approach from commercially available (R)-epichlorohydrins with a 7-step procedure [4f]. The synthesis enables us to obtain sub-gram of AA005 (1) to support a variety of cell-based studies, while, it seems to be tedious, of high labor cost and unsatisfying for scale-up preparation. In this study, we explored a shortcut for rapid assembly of the unique fragments, re-organized and optimized the order of reactions, and eventually completed a multi-gram synthesis of AA005 (1) in ten overall steps from the simple starting materials.
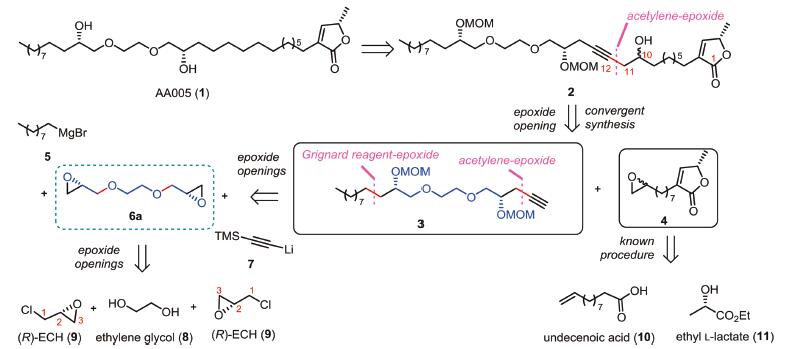
It was envisioned that AA005 (1) might arise from the internal alkyne 2 through a series of functional groups manipulations (Scheme 1). Devising a hydroxyl group at C10 of 2 not only helps the further disconnection of C11-C12 bond through epoxide-opening protocol with a terminal alkyne intermediate 3, but also leaves us an anchor to link labeling functionalities with AA005 (1) in further biological studies [4e]. It is noteworthy that the absolute stereochemistry of the C10 hydroxyl group of 2 could be neglected in the synthesis of AA005 (1) [4c]. The resulting epoxide 4 could be readily traced to abundant materials undecenoic acid (10) and ethyl L-lactate (11) through a key 3-step procedure [4d]. Utilizing partial C2 symmetry at the middle region, disconnection converts the polyether segment 3 into building blocks 5, 6a and 7. The C2 symmetrical bis-epoxide 6a, in principle, would be accessible from the commercially available glycol (8) and (R)-epichlorohydrin (9, abbrev. as (R)-ECH in the following text) [7, 8]. Such an expeditious access could create the requisite polyether unit bearing two stereogenic cabinols in a more direct fashion, and thus shortens the overall steps of the synthesis. However, such a direct transformation has to check the associated regioselectivity with the two possible nucleophilic attacks on epoxide 9 with glycol 8. If random attacks happens upon either carbons of C1-Cl bond and epoxide C3-O bond of (R)-ECH (9) by glycol 8, the reaction would deliver bis-epoxide 6a with poor enantiomeric purity finally.

|
Download:
|
| Scheme 1. Retrosynthetic analysis of AA005 (1). | |
{kind=link}
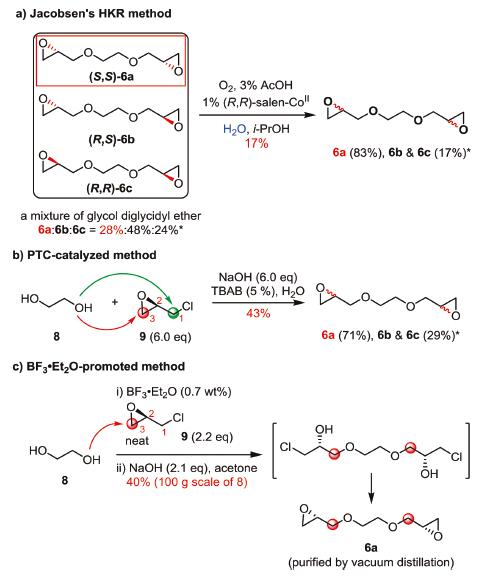
As mentioned above, the newly designed synthesis of AA005 (1) is highly depended on the preparation of enantiopure bis-epoxide 6a, as well as its scalability. To clarify these concerns, three mechanism-different approaches were examined at the first stage (Scheme 2). One is the hydrolytic kinetic resolution (HKR), which has emerged as a widely adopted powerful method to produce various terminal epoxides with high enantiopurities [9]. A mixture of 6 (containing 6a/6b/6c) was applied as the substrate and chiral SalenCoIII complex was used as the catalyst in the assessment (Scheme 2a). The chiral HPLC analysis of the starting glycol diglycidyl ether 6 (industrial grade) showed that it comprises (S,S)-6a, meso-(R,S)-6b and (R,R)-6c with a ratio of 28:48:24. Under the standard conditions with 1 mol% (R,R)-Salen-CoIII·OAc prepared in situ [9b], the percentage of 6a in the mixture increased up to 83% (30% yield based on 52% content of 6a/6c in the mixture; 17% isolated yield based on all the material). Such a product purity was insufficiently qualified for the further synthesis. Phase transfer catalyst (PTC) approach was next examined by nucleophilic attack of glycol (8) on (R)-ECH (9) under basic conditions [7]. Nevertheless, it was difficult to predict, a priori, which carbon (C1-Cl in green, or C3-O in red) of 9 would be predominately attacked by the alkoxy anion of 8. HPLC measurement showed that this approach could not deliver a good result either (Scheme 2b), and all the 6a, 6b (the diastereomer of 6a) and 6c (the enantiomer of 6a) existed in the mixture with a ratio of 71:26:3 after the reaction. Several commonly available PTCs, including TBAB, TBAI and TEBAC, were tested to improve the regioselectivity, as well as varying base concentrations. Unfortunately, all these attempts delivered similar results. Obviously, both C1 and C3 of epoxide 9 were attacked by glycol 8 under the PTC conditions. The Lewis acid-promoted conditions were finally explored (Scheme 2c) [8]. To facilitate future scale-up, the neat reaction (without solvent) was tried [8b]. To our delight, a bis-chlorohydrin intermediate was detected when a mixture of diol 8 and BF3·Et2O (0.7 wt%) was exposed to epoxide 9. It indicated that nucleophilic attack of diol 8 on 9 takes place only at C3-O of epoxide 9 in the presence of BF3·Et2O. To simplify the procedure, the above resulting bis-chlorohydrin intermediate (after removal of excess (R)-ECH (9) in vacuum) was immediately treated with NaOH (2.1 equiv.) to rebuild the two epoxides [10], affording enantiopure (S,S)-6a (for details of condition screening see Supporting information). In a scale-up experiment (using 100 g of ethylene glycol 8 as raw material), approximately 111 g of 6a could be harvested in one batch after final purification by vacuum distillation. Obviously, this reaction shows great scalability and advantages of using economically available materials, ease of operation, convenient purification and mild reaction conditions.

|
Download:
|
| Scheme 2. Three individual approaches explored to prepare bis-epoxide 6a. *The product ratio was determined by chiral HPLC analysis (LC-20AP, 2*AD-H, RID, n-hexane/i-PrOH (95/5), flow rate of 1.0 mL/min). | |
{kind=link}
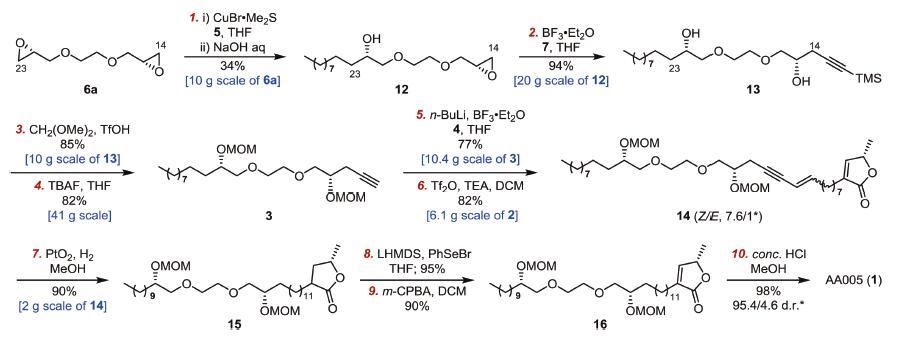
With large quantity of the C2-symmetric bis-epoxide 6a in hand, subsequent C23- and C14-functionalizations of 6a were executed successively (Scheme 3). Grignard addition of 6a (10 g, 57 mmol) with 1.2 equiv. of nonylmaganesium bromide 5 in the presence of CuBr·Me2S (20 mol%) at -78 ℃ followed by treatment with NaOH (0.5 equiv.) afforded the mono-epoxide 12 (6 g, 34% yield after recrystallization) [11], while the optimized reaction could proceed in 60% yield in a smaller scale of 2.8 mmol of 6a (Supporting information for more details). The following five steps were carried out smoothly to construct the whole skeleton of AA005 (1). Epoxide 12 (20 g) was treated with an excess of lithium acetylides 7 in the presence of BF3·Et2O [12], affording the ring-opened adduct 13 in 94% yield. After MOM protection of the two secondary alcohols of 13 (10 g) [13], desilylation of the resulting compound (41 g) with TBAF gave the terminal alkyne 3 in 82% yield. The other lactone-epoxide segment 4, which was prepared from the known procedure [4d], was subjected to the coupling with lithium acetylides of 3 (10.4 g) in the presence of BF3·Et2O, delivering the whole skeleton 2 in 77% yield. The free alcohol 2 (6.1 g) was then dehydrated by treatment with Tf2O and TEA [14], giving the conjugated enyne 14 in 82% yield.

|
Download:
|
| Scheme 3. Synthesis of AA005 (1) via enyne 14. *The Z/E ratio of 14 was determined by 1H NMR. And the dr value at C34 of AA005 (1) was determined by chiral stationary phase HPLC analysis (Chiralpak® AS-H, DAD (detector), mobile-phase (n-hexane/i-PrOH (90/10)), flow rate of 0.5 mL/min). | |
{kind=link}
From enyne 14 to the completion of the synthesis of AA005 (1), the remaining steps include chemoselective hydrogenation of unsaturated bonds and global removal of the protecting groups. Unfortunately, attempts to directly convert 14 into 16 encountered a tricky problem of over-reduction of 14. Either diimide reduction [15] or Wilkinson homogeneous hydrogenation [16] was proven to be fruitless, affording 15 and 16 as an inseparable mixture. To circumvent the above problem, a detour was adopted from enyne 14 (Scheme 3). Complete reduction of unsaturated bonds of 14, followed by phenylselenide introduction and oxidative elimination rebuilt the conjugated C=C bond [17], afforded the single product 16. Finally, global cleavage of the MOM protecting groups was carried out under acidic conditions, furnishing AA005 (1, dr = 95.4:4.6 analyzed by chiral HPLC) in 98% yield. Such a manner avoided the above-mentioned problem caused by unsatisfying regioselective hydrogenation, however, its poor redox and step economies and use of toxic phenylselenide reagent hinder the scale-up capability of the synthesis.
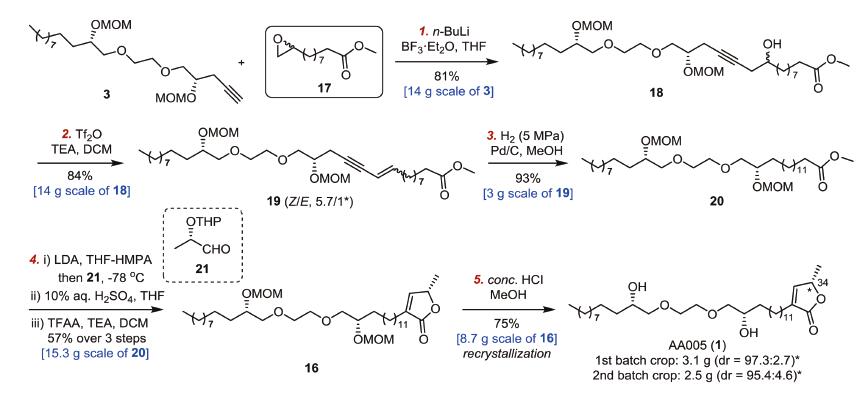
We decided to reorganize the reaction sequence and postpone the introduction of the unsaturated lactone functionality in the synthesis. To circumvent the chemoselectivity of hydrogenation, epoxide-ester 17 (prepared from undecenoic acid 10 [18], to displace epoxide-lactone 4) was applied for the fragment coupling with terminal alkyne 3 (Scheme 4). Accordingly, formation of the butenolide functionality was adjusted to the final stage in the synthesis. Such a change may restore the redox economy in the scalable synthesis. Coupling of terminal acetylene 3 (14 g) with epoxide 17 afforded alcohol 18, which underwent dehydration to yield the conjugated enyne 19 in gram quantities. Full hydrogenation of enyne 19 (3 g) was carried out under hydrogen atmosphere (5 MPa) in the presence of a catalytic amount of 10% Pd/C, giving the corresponding saturated ester 20 in 93% yield. The aldol condensation of in situ generated enolate of 20 (15.3 g) with aldehyde 21 (readily prepared from ethyl L-lactate (11) [4d]) followed by treatment with 10% aq. H2SO4 in THF led to selective cleavage of THP protecting group and subsequent closure of the lactone. Exposure of the resulting mixture of β-hydroxy-γ-lactones to TFAA and TEA trigged β-elimination of trifluoroacetic acid (TFA) and assembled the whole skeleton 16 (57% yield over 3 steps from 20). Finally, global deprotection under conc. HCl in methanol delivered AA005 (1, 5.6 g) in 75% yield after recrystallization. The spectroscopic properties of synthetic AA005 (1) (1H, 13C NMR, and [α]D20 were in well agreement with those reported [4a]. Moreover, the dr (partial epimerization of the allylic H/Me at C34 [6b]) was also measured and confirmed by chiral HPLC analysis.

|
Download:
|
| Scheme 4. Completion of the gram-scale laboratory synthesis of AA005 (1) via enyne 19.*The Z/E ratio of 19 was determined by 1H NMR. The dr value at C34 of AA005 (1) was determined by chiral stationary phase HPLC analysis (Chiralpak® AS-H, DAD (detector), mobile-phase (n-hexane/i-PrOH (90/10)), flow rate of 0.5 mL/min). | |
{kind=link}
In summary, we have successfully accomplished a scalable synthesis of antitumor ACGs mimic AA005 (1), which is capable to render the final product in multiple grams with high enantiomeric and diastereomeric purity from economically available starting materials. This ten-step laboratory synthesis of AA005 (1) features with application of (a) scalable BF3·Et2O-promoted epoxide-opening of (R)-epichlorohydrin with glycol to generate bis-epoxide 6a in one pot, (b) quick construction of the whole skeleton with successive epoxide-openings of bis-epoxide 6a, and (c) late-stage formation of the butenolide to avoid the chemoselectivity of hydrogenation in the synthesis. Optimal combination of these methodologies and reaction sequence led to satisfying step and redox economies. We believe that the disclosed scalable synthesis of AA005 is capable to support current biological investigations, including ongoing animal experiments. Furthermore, availability of large quantity of late-stage intermediates from this new synthesis enables us to elaborate various molecular tools of AA005 for the mechanism studies.
Declaration of competing interestThe authors declare no competing financial interest.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (Nos. 21532002, 21761142001 and 21778031) and National Key Research and Development Program (No. 2018YFC0310900) is greatly appreciated.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.06.007.
| [1] |
(a) A. Bermejo, B. Figadere, M.C. Zafra-Polo, et al., Nat. Prod. Rep. 22 (2005) 269-303; (b) T.S. Hu, Y.L. Wu, Z.J. Yao, Recent progress on the chemical synthesis of Annonaceous acetogenins and their structurally modified mimics, in: Y.L. Wu, Z.J. Yao (Eds.), Synthesis of Natural Products: A Scientific and Artistic Exploration, Science Press, Beijing, 2006, pp. 58-90; (c) J.L. McLaughlin, J. Nat. Prod. 71 (2008) 1311-1321; (d) S. Gajalakshmi, R. Divya, V. Divya-Deepika, et al., Int. J. Pharm. Sci. Rev. Res. 10 (2011) 24-29; (e) S.M. Agarwal, M.I. Khan, M. Mangal, Anticancer Agents Med. Chem. 16 (2016) 138-159. |
| [2] |
D. Cortes, B. Figadere, A. Cave, Phytochemistry 32 (1993) 1467-1473. DOI:10.1016/0031-9422(93)85161-J |
| [3] |
(a) S. Hoppen, U. Emde, T. Friedrich, et al., Angew. Chem. Int. Ed. 39 (2000) 2099-2102; (b) T. Gallardo, M.C. Zafra-Polo, J.R. Tormo, et al., J. Med. Chem. 43 (2000) 4793-4800; (c) S. Arndt, U. Emde, S. Bäurle, et al., Chem. Eur. J. 7 (2001) 993-1005; (d) N. Kojima, T. Tanaka, Molecules 14 (2009) 3621-3661; (e) T. Matsumoto, N. Kojima, A. Akatsuka, et al., Tetrahedron 73 (2017) 2359-2366. |
| [4] |
(a) B.B. Zeng, Y. Wu, Q. Yu, et al., Angew. Chem. Int. Ed. 39 (2000) 1934-1937; (b) S. Jiang, Z.H. Liu, G. Sheng, et al., J. Org. Chem. 67 (2002) 3404-3408; (c) B.B. Zeng, Y. Wu, S. Jiang, et al., Chem. Eur. J. 9 (2003) 282-290; (d) S. Jiang, Y. Li, X.G. Chen, et al., Angew. Chem. Int. Ed. 43 (2004) 329-334; (e) H.X. Liu, G.R. Huang, H.M. Zhang, et al., ChemBioChem 8 (2007) 172-177; (f) Q.C. Xiao, Y.Q. Liu, Y.T. Qiu, et al., J. Med. Chem. 54 (2011) 525-533; (g) B. Han, Y.X. Cao, Z.M. Li, et al., Acta Pharmacol. Sin. 40 (2019) 231-242. |
| [5] |
(a) J.D. White, T.C. Somers, G.N. Reddy, J. Org. Chem. 57 (1992) 4991-4998; (b) Z.J. Yao, Y.L. Wu, Tetrahedron Lett. 35 (1994) 157-160; (c) Z.J. Yao, Y.L. Wu, J. Org, Chem. 60 (1995) 1170-1176. |
| [6] |
(a) P. Duret, B. Figadere, R. Hocquemiller, A. Cave, Tetrahedron Lett. 38 (1997) 8849-8852; (b) Q. Yu, Y.K. Wu, Y.L. Wu, et al., Chirality 12 (2000) 127-129. |
| [7] |
(a) X.P. Gu, I. Ikeda, M. Okahara, Synthesis 16 (1985) 649-651; (b) Z.G. Tang, C.W. Zhang, Y.L. Yue, Huaxue Shiji 2 (1994) 107-109; (c) Q.G. Huang, Guangxi Chem. Ind. 3 (1996) 13-14; (d) Z.X. Liu, L. Wang, C.Y. Bao, et al., Biomacromolecules 12 (2011) 2389-2395. |
| [8] |
(a) D.J. Yue, Hubei Chem. Ind. 1 (1994) 19-21; (b) Y. Zhang, Y.S. Xu, S.J. Qiu, L. Yang, Chem. Ind. & Eng. 31 (2014) 31-36. |
| [9] |
(a) M. Tokunaga, J.F. Larrow, F. Kakiuchi, E.N. Jacobsen, Science 277 (1997) 936-938; (b) S.E. Schaus, B.D. Brandes, J.F. Larrow, et al., J. Am. Chem. Soc. 124 (2002) 1307-1315; (c) L.P.C. Nielsen, C.P. Stevenson, D.G. Blackmond, E. N. Jacobsen, J. Am. Chem. Soc. 126 (2004) 1360-1362. |
| [10] |
F.D. Liu, K.K. Guo, J.M. Yuan, High Perform. Polym. 26 (2014) 326-334. DOI:10.1177/0954008313514083 |
| [11] |
(a) A. Furstner, M. Albert, J. Mlynarski, M. Matheu, E. DeClercq, J. Am. Chem. Soc. 125 (2003) 13132-13142; (b) R. Schmidt, M. Ostermeier, R. Schobert, J. Org. Chem. 82 (2017) 9126-9132. |
| [12] |
M. Yamaguchi, I. Hirao, Tetrahedron Lett. 24 (1983) 391-394. DOI:10.1016/S0040-4039(00)81416-1 |
| [13] |
M.P. Groziak, A. Koohang, J. Org. Chem. 57 (1992) 940-944. DOI:10.1021/jo00029a027 |
| [14] |
T. Durand, A. Guy, J.P. Vidal, J.C. Rossi, J. Org. Chem. 67 (2002) 3615-3624. DOI:10.1021/jo0109624 |
| [15] |
(a) D.J. Hart, W.P. Hong, L.Y. Hsu, J. Org. Chem. 52 (1987) 4665-4673; (b) J.A. Marshall, M.Z. Chen, J. Org. Chem. 62 (1997) 5996-6000. |
| [16] |
(a) T.R. Hoye, Z.X. Ye, J. Am. Chem. Soc. 118 (1996) 1801-1802; (b) D.J. Dixon, S.V. Ley, D.J. Reynolds, Angew. Chem. Int. Ed. 39 (20003622-3626; (c) S. Takahashi, A. Kubota, T. Nakata, Angew. Chem. Int. Ed. 41 (2002) 4751-4754. |
| [17] |
J.K. Fu, H.J. Shen, Y.Y. Chang, et al., Chem. Eur. J. 20 (2014) 12881-12888. DOI:10.1002/chem.201403756 |
| [18] |
(a) T. Vijai-Kumar-Reddy, B.L. A. Prabhavathi-Devi, R.B.N. Prasad, P. Sujitha, C. Ganesh-Kumar, Eur. J. Med. Chem. 67 (2013) 384-389; (b) Y.J. Chen, S. Jin, J. Xi, Z.J. Yao, Tetrahedron 70 (2014) 4921-4928. |