 2021, Vol. 32
2021, Vol. 32 


b Department of Applied Chemistry, K. N. Toosi University of Technology, Tehran, Iran
Palladium-catalyzed decarboxylative cycloaddition reaction has become an efficient method for the synthesis of N-heterocycles since Tunge firstly applied vinyl benzoxazinanones as a powerful precursor [1]. A variety of [4 + 1] [2], [4 + 2] [3] and [4 + 3] [4] reactions have been developed to prepare 5-, 6- and 7-membered nitrogen-containing compounds diastereo- and enantioselectively via the zwitterionic intermediates after oxidative addition/ decarboxylation with metal complexes. α, β-Unsaturated carbonyl compounds, sulfur-ylide, ketene and many other reagents were utilized to introduce the functional group complexity (Scheme 1, eq. 1). Nucleophilic addition to the more-substituted C1 carbon in the π-allyl/Pd intermediate was proposed. On the other hand, stoichiometric [5] and catalytic [6] cyclopropane formation reactions have been developed from allylic substrates by thenucleophilic attack to C2 carbon in the π-allyl/Pd complex via palladacyclobutane intermediate. However, reaction involving the palladacyclobutane intermediate from vinyl benzoxazinanone precursor is still very rare. Zhai reported an elegant tetracyclic indoline synthesis, in which the migratory insertion of the palladacyclobutane to benzyne intermediate was proposed as the possible mechanism (Scheme 1, eq. 2) [7, 8]. We assume that the palladacyclobutane species can undergo β-hydride elimination when R is methyl group and the stereocenter at C2 can be controlled by chiral ligand in the C—N bond formation step. Herein, we report the Pd-catalyzed enantioselective synthesis of 2-methyl-3-methyleneindoline [9] in up to 89% yield and 84% ee with (R,R)-BenzP* as the chiral ligand.

|
Download:
|
| Scheme 1. Reaction pathways for zwitterionic intermediate from vinyl benzoxazinanone. | |
We started our study with methyl-substituted vinylbenzoxazinanone 1a as the model substrate (Table 1).
|
|
Table 1 Optimization of reaction conditions.a |

{kind=link}
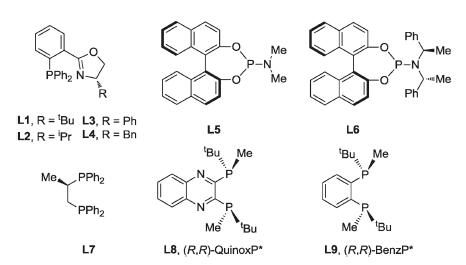
Different chiral ligands (Fig. 1) were examined using 5 mol% Pd(dba)2 in THF at 80 ℃. Reactions in the presence of chiral Phox ligands with different substitutions at the oxazoline ring (L1-L4) gave moderate to high yields of 2a, while up to 60% ee could be observed (entries 1–4). The enantioselecltivities with different chiral phosphoramidite ligands are also low (53% and 30%), although the yields of 2a are high (entries 5 and 6). A variety of chiral diphosphine ligands were evaluated and higher chemical yields of 2a could be obtained when L5-L7 with two-carbon linker between the phosphine atoms are used. The highest enantioslectivity is 80% in case (R,R)-BenzP* was applied (entry 9). 2a with 82% ee was synthesized when the reaction was conducted at 70 ℃. Remarkable solvent effect was observed. Reactions in dioxane and DCE could not further improve the enantioselectivities (entries 11 and 12). Reaction in DMF affords 2a with 86% ee, while the yield dropped to 30% (entry 13). Finally, reaction in toluene at 70 ℃ gave 89% of 2a in 82% ee, which was selected as the optimized condition (entry 14).

|
Download:
|
| Fig. 1. Structures of chiral ligands. | |
{kind=link}
With the optimized conditions in hand, we investigated the scope of the asymmetric synthesis of 2-methyl-3-methyleneindoline (Scheme 2). Chlorides at 6 or 7 position in the substrates have little effect to the selectivities (2b and 2c). On the contrast, the reaction of 1d with a chloride at 5 position is sluggish. 2d could be isolated in 60% yield and the enantioselectivity dropped to 68% dramatically (2d). Electron-withdrawing bromide and fluoride (2e and 2f) could be tolerated under the optimized condition as well as electron-donating methyl and methoxy groups (2g and 2h). The presence of alkyne function in the substrate lead to lower conversion, while the enantioselectivity remains to be 82% (2i). Substrates with aromatic phenyl, 2-naphthyl, 3-furanyl groups at 6-position could also react smoothly to give the 2-methyl-3-methyleneindoline in high yields but relatively lower enantioselectivities (2j, 2k and 2l). Besides the substituents on the aromatic ring, the influence of the protecting groups on the nitrogen was also examined. The same level of enantioselectivities was obtained when simple methyl, phenyl, 4-MeOC6H4 or 4-FC6H4 sulfonyl groups were used (2m to 2p). However, substrate with Ac groups leads to low conversion. When 1q with an ethyl group was subjected to the same reaction condition, 2q was isolated in 65% yield with E configuration, while only 20% ee was observed, probably due to the relatively slow interconversion of the racemic substrates. Substitutions on the vinyl group lead to complicated reaction mixture formation. The absolute configuration of 2a was assigned to be R by single crystal X-ray diffraction analysis.

|
Download:
|
| Scheme 2. The scope of the Pd-catalyzed 2-methyl-3-methyleneindoline synthesis. | |
{kind=link}
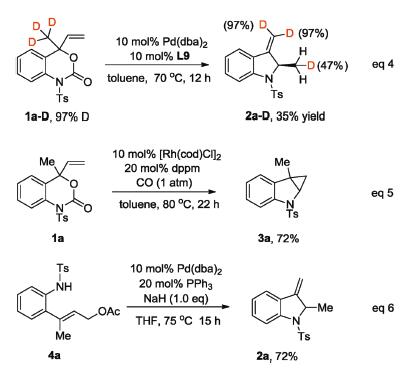
Several experiments were conducted to investigate whether the palladacyclobutane formation and β-hydride elimination mechanism is involved in the reaction (Scheme 3). Firstly, 1a-D with deuterated methyl group could be transformed to 2a-D in 35% yield under the identical condition, in which one of the hydrogen in the methyl group was deuterated in 43% (eq. 4). This reaction indicates that β-hydride elimination occurs in this transformation and it is likely a slow step in the catalytic cycle. Secondly, the reaction of 1a with Pd performed in the atmosphere of CO failed to give any cyclobutanone product as expected. However, 3a with a cyclopropane skeleton was isolated in 72% yield when [Rh(cod)Cl]2 and dppm were used as the catalyst (eq. 5). Thirdly, the dynamic kinetic asymmetric synthesis of 2a from racemic 1a suggest a fast π-σ-π equilibrium exists in the catalysis, which was supported by the fact that 4a synthesized independently could be transformed into the same product 2a with Pd under basic condition (eq. 6).

|
Download:
|
| Scheme 3. Mechanism studies. | |
{kind=link}
Based on the experiments above and literature reports, we proposed a plausible mechanism as shown in Scheme 4. 1a undergoes oxidative addition with Pd complex and releases CO2 to form zwitterionic intermediate A. A could have a fast equilibrium with σ-allyl Pd complex D to realize the interconversion of the diastereomeric intermediate. The linear substrate 4a can be converted to the same complex A via D. From A, intramolecular attack of nitrogen to C3 leads to the formation of byproduct. While the attack to C2 generates the palladacyclobutane intermediate B, which is the stereo-determine step. Product 2a could be obtained from B via β-hydride-elimination and reductive elimination sequence. The direct reductive elimination from B to give cyclopropane 3 is not observed for palladium. While 3 was isolated with Rh catalyst in the presence of CO atmosphere.

|
Download:
|
| Scheme 4. The proposed reaction mechanism. | |
{kind=link}
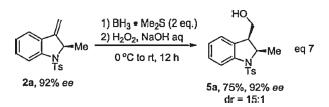
2a with 92% ee could be obtained by a simple recrystallization. A further hydroboration/oxidation sequence converts the double bond to the hydroxy group diastereoselectively to give 5a in 75% yield and 15 : 1 dr (eq. 7).
|
In summary, we have reported an enantioselective Pd-catalyzed 2-methyl-3-methyleneindoline synthesis from racemic vinyl benzoxazinanones with the help of (R,R)-BenzP*. Mechanism studies support a C2 attack to π-allyl Pd complex and the formation of palladacyclobutane as the key intermediate. The β-hydride elimination provides a new reaction pathway besides migratory insertion to benzyne, which inspires us to explore further transformation for the palladacyclobutane structure.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is supported by National Natural Science Foundation of China (NSFC, No. 21602130), and Shanghai Jiao Tong University.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.02.056.
| [1] |
(a) C. Wang, J.A. Tunge, Org. Lett. 8 (2006) 3211-3214; (b) C. Wang, J.A. Tunge, J. Am. Chem. Soc. 130 (2008) 8118-8119. |
| [2] |
(a) T.R. Li, F. Tan, L.Q. Lu, et al., Nat. Commun. 5 (2014) 5500; (b) Q. Wang, X. Qi, L.Q. Lu, et al., Angew. Chem. Int. Ed. 55 (2016) 2840-2844. |
| [3] |
(a) C. Wang, N. Pahadi, J.A. Tunge, Tetrahedron 65 (2009) 5102-5109; (b) G.J. Mei, C.Y. Bian, G.H. Li, et al., Org. Lett. 19 (2017) 3219-3222; (c) L.A. Leth, F. Glaus, M. Meazza, et al., Angew. Chem. Int. Ed. 55 (2016) 15272-15276; (d) Y. Wei, L.Q. Lu, T.R. Li, et al., Angew. Chem. Int. Ed. 55 (2016) 2200-2204; (e) M.M. Li, Y. Wei, J. Liu, et al., J. Am. Chem. Soc. 139 (2017) 14707-14713. |
| [4] |
(a) C. Guo, M. Fleige, D. Janssen-Müller, C.G. Daniliuc, F. Glorius, J. Am. Chem. Soc. 138 (2016) 7840-7843; (b) C. Guo, D. Janssen-Müller, M. Fleige, et al., J. Am. Chem. Soc. 139 (2017) 4443-4451. |
| [5] |
(a) L.S. Hegedus, W.H. Darlington, C.E. Russell, J. Org. Chem. 45 (1980) 5193-5196; (b) H.M.R. Hoffmann, A.R. Otte, A. Wilde, S. Menzer, D.J. Williams, Angew. Chem. Int. Ed. 34 (1995) 100-102. |
| [6] |
(a) A. Satake, T. Nakata, J. Am. Chem. Soc. 120 (1998) 10391-10396; (b) A. Satake, H. Kadohama, H. Koshino, T. Nakata, Tetrahedron Lett. 40 (1999) 3597-3600; (c) R. Shintani, S. Park, T. Hayashi, J. Am. Chem. Soc. 129 (2007) 14866-14867; (d) W. Liu, D. Chen, X.Z. Zhu, X.L. Wan, X.L. Hou, J. Am. Chem. Soc. 131 (2009) 8734-8735; (e) R. Shintani, T. Tsuji, S. Park, T. Hayashi, J. Am. Chem. Soc. 132 (2010) 7508-7513; (f) R. Shintani, K. Moriya, T. Hayashi, Chem. Commun. 47 (2011) 3057-3059; (g) R. Shintani, T. Ito, M. Nagamoto, H. Otomo, T. Hayashi, Chem. Commun. 48 (2012) 9936-9938; (h) R. Shintani, T. Ito, T. Hayashi, Org. Lett. 14 (2012) 2410-2413; (i) J.Q. Huang, C.H. Ding, X.L. Hou, J. Org. Chem. 79 (2014) 12010-12017; (j) J.Q. Huang, W. Liu, B.H. Zheng, et al., ACS Catal. 8 (2018) 1964-1972; (k) G.P. Zhang, S. Huang, Y.J. Jiang, et al., Chem. Commun. 55 (2019) 6449-6452. |
| [7] |
S. Duan, B. Cheng, X. Duan, et al., Org. Lett. 20 (2018) 1417-1420. DOI:10.1021/acs.orglett.8b00192 |
| [8] |
N. Punna, K. Harada, J. Zhou, N. Shibata, Org. Lett. 21 (2019) 1515-1520. DOI:10.1021/acs.orglett.9b00330 |
| [9] |
(a) R.C. Larock, J.M. Zenner, J. Org. Chem. 60 (1995) 482-483; (b) J.M. Zenner, R.C. Larock, J. Org. Chem. 64 (1999) 7312-7322; (c) W. Shu, Q. Yu, S. Ma, Adv. Synth. Catal. 351 (2009) 2807-2810. |