 2021, Vol. 32
2021, Vol. 32 


b School of Pharmaceutical Sciences and the Innovative Drug Research Centre, Chongqing University, Chongqing 401331, China
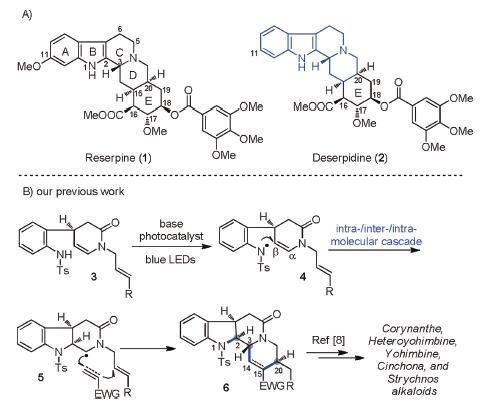
Reserpine (1) and its congener deserpidine (2) are two prominent Rauwolfia alkaloids with potent antihypertensive and sedative activities (Scheme 1A) [1]. Since they were isolated in 1950s [2], they have attracted considerable attention and been extensively explored on structural elucidation [3], synthesis [4-6] and pharmacological properties [7]. Structurally, reserpine and deserpidine only differ in the C11 substituent. Both of them feature the yohimbine framework with six chiral centers and a densely substituted cyclohexane E ring embedded in the pentacyclic ring system, which poses a synthetic challenge. Reserpine has been considered as one of the most historic targets for synthetic community. Since the pioneering total synthesis of reserpine was accomplished by Woodward in 1956 [5a, b], diverse synthetic strategies have emerged [5c-l].

|
Download:
|
| Scheme 1. (A) Structures of reserpine and deserpidine. (B) Total synthesis of monoterpenoid indole alkaloids via intra-/inter-/intra- molecular photocatalytic radical cascade reaction developed by our group. | |
{kind=link}
Compared with reserpine, total synthesis of deserpidine was seldom reported [6], and the only asymmetric one relied on the conversion of reserpine to deserpidine [6f]. Although deserpidine has been used in the treatment of hypertension and psychosis [7d-f], its poor availability from natural sources (only 0.04% from the extracts of Rauwolfia canescens) resulted in limited clinical applications. Therefore, it is necessary to develop an efficient strategy to synthesize deserpidine and its derivatives for the further exploration of their biological activities.
In continuation of our long-standing interest in total synthesis of complex indole alkaloids, our group has been focused on developing unified synthetic methodologies which could enable the rapid assembly of intricate molecular architectures and diverse synthetic targets. Recently, we have disclosed three types of visible-light photocatalytic radical cascade reactions triggered by N-centered radicals, providing efficient access to the asymmetric total synthesis of alkaloids with tetrahydrocarbolinone ring systems [8]. Among them, as depicted in Scheme 1B, the intra-/inter-/intramolecular type cascade reaction between sulfoanimde 3 bearing unsaturated side chains and external Michael acceptors was successfully realized to build the tetracyclic corynanthe core with N1—C2, C3—C14, and C15—C20 bonds along with C2, C3, C20 stereocenters in one-pot, leading to the total synthesis of a series of corynanthe, heteroyohimbine, yohimbine, strychnos alkaloids [8]. In contemplating an expedient synthetic approach to deserpidine containing the corynanthe scaffold, we envisioned that assembly of the ABCD ring system via the above intra-/inter-/intramolecular radical cascade reaction followed by the formation of ring E would be applicable. Herein, we reported our synthetic efforts toward the asymmetric total synthesis of deserpidine.
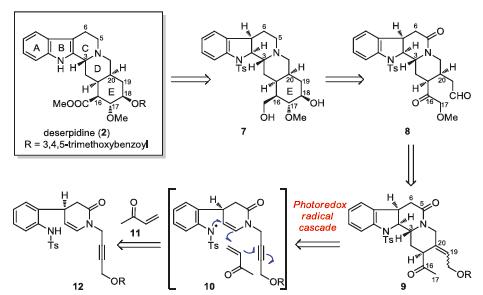
As illustrated retrosynthetically in Scheme 2, deserpidine could be derived from compound 7 by the late-stage functional group manipulations. Bond disconnection of the E ring in 7 led back to aldehyde 8. Next, compound 9 with a primary alcohol functionality was recognized as the appropriate intermediate for approaching 8. Finally, construction of key tetracyclic intermediate 9 by visible light-induced radical cascade reaction (10) unveiled propargyl alcohol 12 and methyl vinyl ketone 11 as the reaction substrates.

|
Download:
|
| Scheme 2. Retrosynthetic analysis of deserpidine. | |
{kind=link}
Our synthesis commenced with the preparation of the radical precursor 16 for photocatalytic cascade reaction (Scheme 3). First, condensation of 4-benzyloxyl propargyl amine 14 [9] with the known chiral aldehyde 13 proceeded smoothly in the presence of AcOH in refluxing toluene to provide enamide 15 (78%) [9]. After reduction of the nitro group in 15, tosyl (Ts) protection of the resulting amine afforded the key substrate 16 in 55% yield. On the basis of our previous research [8, 9], the designed intra-/inter-/intramolecular radical cascade reaction was performed upon treating 16 and methyl vinyl ketone 11 with blue LED irradiation under the conditions of Ir(dtbbpy)(ppy)2PF6/KHCO3/THF. As expected, the desired tetracyclic product 17 was obtained as a mixture of diastereomers at C15 and geometrical isomers at C19 in 60% overall yield (2:1.8:1.2:1 dr) with the corynanthe scaffold assembled. It is worth mention that the stereochemistry of the newly formed C2 and C3 was in full control. After the intensive investigation of the reaction conditions, we found that decreasing the concentration of substrate 16 in the radical reaction from 20 μmol/mL to 7 μmol/mL significantly improved the yield (from 60% to 93%), which insured the high efficiency of the above conversion. In order to reduce the complexity caused by the geometric isomerism at C19 and make the full use of the four diastereomers, it is necessary to migrate the exocyclic double bond in 17 into the D ring. Accordingly, after installation of the requisite methoxy group of deserpidine at C17 in 17 by using PhI(OAc)2/BF3﹞Et2O in methanol [10], the resulting compound 18 was directly treated with DBU to generate 19 as a pair of inseparable diastereomers in 50% overall yield with a 5:1 dr at C15. Next, 19 was subjected to catalytic hydrogenation. At this stage, compound 20 derived from the major diastereoisomer of 19 was separated as a single isomer in 80% yield (see Supporting information for details) while the reduction product from the minor one of 19 was inseparable with some uncertain byproducts (12% yield). Oxidation of alcohol 20 to aldehyde 21 and subsequent intramolecular aldol reaction prompted by DBU forged the C17—C18 bond, providing pentacyclic compound 22 as a pair of inseparable diastereomers (6:1) in 50% yield over two steps. To determine the configuration of 22, the single crystal of the major isomer 22a was cultivated from a mixture of dichloromethane and methanol. The absolute configuration of 22a was established unequivocally as 15S, 17R, 18R and 20R by X-ray crystallographic analysis.

|
Download:
|
| Scheme 3. Preparation of pentacycle 22. | |
{kind=link}
With the pentacyclic compound 22 in hand, we began to explore the late-stage synthesis of the stereoisomer of deserpidine. As shown in Scheme 4, we first attempted to convert ketone 22 into its homologous nitrile 24 by van Leusen reaction [11]. We envisaged that the cyano group in 24 could be easily transformed into the methyl ester group at C16 in deserpidine. However, under the conditions of TosMIC/t-BuOK/DME/EtOH at 25 ℃, we only detected the production of intermediate 23 (24% yield) along with some other unknown side-products. Even though different temperatures and solvents of the reaction were screened, 23 was not able to undergo further transformation to give the desired nitrile 24. Therefore, we turned to perform Corey-Chaykovsky reaction of the inseparable mixture 22 by using Me3SI and t-BuOK, which gave rise to epoxide 25 as the main product in 72% yield, and the structure of 25 was unambiguously elucidated by X-ray crystallography. Subsequent reductive ring-opening of epoxide in 25 with Zn/Cp2TiCl2 furnished 26a and 26b as a pair of separable diastereomers in the yields of 65% and 23%, respectively. We envisioned that both 26a and 26b could be carried forward to prepare the derivatives of deserpidine. Therefore, compound 26a was converted into the corresponding methyl ester via oxidation of the alcohol into aldehyde, Pinnick oxidation and subsequent esterification with TMSCHN2 in methanol, providing 27 in 63% yield over three steps. After removing the Ts group of 27 with Mg/MeOH, the resultant aniline was oxidized with (PhSeO)2O to produce indole 28. Finally, further reduction of the amide in 28 with [Rh(H)CO](PPh)3/PhSiH3 in anhydrous THF followed by introduction of the 3, 4, 5-trimethoxylbenzoyl group [5i, 6f] on the hydroxyl group at C18 afforded the 16, 17, 20-epi-deserpidine (29) in 69% overall yield. Meanwhile, we investigated the transformation of the minor diastereomer 26b. When 26b was oxidized under the conditions of TEMPO/PhI(OAc)2/CH2Cl2, the resulting aldehyde intermediate spontaneously reacted with C18 hydroxyl group and underwent further oxidation, thus delivering lactone 30 in 86% yield. Then, lactone 30 was elaborated into indole 31 in 50% overall yield via a three-step synthetic sequence involving the removal of Ts group, oxidation of aniline, and reduction of amide. After much experimentation, we failed to open the lactone ring in 31, revealing that the lactone functionality was stable.

|
Download:
|
| Scheme 4. Synthesis of the 16, 17, 20-epi-deserpidine 29 and lactone derivative 31 of (-)-deserpidine. | |
{kind=link}
In conclusion, we have disclosed an efficient approach of the asymmetric total synthesis of 16, 17, 20-epi-deserpidine (29) and the lactone derivative 31 of (-)-deserpidine, which relied on aphotoredox catalytic intra-/inter-/intramolecular radical cascade reaction to assemble the tetracyclic ring system (17). This synthetic strategy builds a foundation for the synthesis of more derivatives of yohimbine alkaloids.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for the financial surpport from the National Natural Science Foundation of China (Nos. 21702140 and 21732005) and National Science and Technology Major Projects for "Major New Drugs Innovation and Development" (No. 2018ZX09101003-005-004).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.02.009.
| [1] |
R.E. Woodson, H.W. Younken, E. Schlitter, J.A. Schneider, Rauwolfia: Botany, Pharmacognosy, ChemistryandPharmacology, Little, Brown andCo., Boston, 1957.
|
| [2] |
(a) J.M. Müller, E. Schlittler, H.J. Bein, Experientia 8 (1952) 338; (b) E. Schlittler, P.R. Ulshafer, M.L. Pandow, R. Hunt, L. Dorfman, Experientia 11 (1955) 64-65. |
| [3] |
(a) H.B. MacPhillamy, C.F. Huebner, E. Schlittler, A.F.St. André, P.R.J. Ulshafer, J. Am. Chem. Soc. 77 (1955) 4335-4343; (b) P.E. Aldrich, P.A. Diassi, D.F. Dickel, J. Am. Chem. Soc. 81 (1959) 2481-2494; (c) E. Smith, R.S. Jaret, M. Shamma, R.J. Shine, J. Am. Chem. Soc. 86 (1964) 2083-2084; (d) M. Lounasmaa, A. Tolvanen, Heterocycles 23 (1985) 371-375. |
| [4] |
(a) E. Wenkert, L.H. Liu, D.B.R. Johnston, J. Org, Chem. 30 (1965) 722-728; (b) S. Takano, F. Ito, K. Ogasawara, Heterocycles 14 (1980) 453-456; (c) T. Suzuki, A. Tomino, K. Unno, T. Kametani, Chem. Pharm. Bull. Jpn. 29 (1981) 76-81; (d) S. Chao, F.A. Kunng, J.M. Gu, H.L. Ammon, P.S. Mariano, J. Org. Chem. 49 (1984) 2708-2711; (e) E. Wenkert, Heterocycles 21 (1984) 325-329; (f) M.E. Jung, L.A. Light, J. Am. Chem. Soc. 106 (1984) 7614-7618; (g) M. Isobe, N. Fukami, T. Nishikawa, T. Goto, Heterocycles 25 (1987) 521-532; (h) R.P. Polniaszek, R.V. Stevens, J. Org. Chem. 51 (1986) 3023-3027. |
| [5] |
(a) R.B. Woodward, F.E. Bader, H. Bickel, A.J. Frey, R.W. Kierstead, J. Am. Chem. Soc. 78 (1956) 2023-2025; (b) R.B. Woodward, F.E. Bader, H. Bickel, A.J. Frey, R.W. Kierstead, Tetrahedron 2 (1958) 1-57; (c) B.A. Pearlman, J. Am. Chem. Soc. 101 (1979) 6404-6408; (d) P.A. Wender, J.M. Schaus, A.W. White, J. Am. Chem. Soc. 102 (1980) 6157-6159; (e) P.A. Wender, J.M. Schaus, A.W. White, Heterocycles 25 (1987) 263-270; (f) S.F. Martin, H. Rueger, S.A. Williamson, S. Grzejszczak, J. Am. Chem. Soc. 109 (1987) 6124-6134; (g) G. Stork, Pure Appl. Chem. 61 (1989) 439-442; (h) S. Hanessian, J. Pan, A. Carnell, H. Bouchard, L. Lesage, J. Org. Chem. 62 (1997) 465-473; (i) G. Stork, P.C. Tang, M. Casey, B. Goodman, M. Toyota, J. Am. Chem. Soc. 127 (2005) 16255-16262; (j) S.M. Sparks, A.J. Gutierrez, K.J. Shea, J. Org. Chem. 68 (2003) 5274-5285; (k) N.S. Rajapaksa, M.A. McGowan, M. Rienzo, W.N. Jacobsen, Org. Lett. 15 (2013) 706-709; (l) J. Park, D.Y.K. Chen, Angew. Chem. Int. Ed. 130 (2018) 16384-16388. |
| [6] |
(a) C. Szántay, G. Blaskó, K. Honty, et al., Liebigs Ann. Chem. 8 (1983) 1292-1309; (b) S. Sakai, M. Ogawa, Heterocycles 10 (1978) 67-71; (c) O. Miyata, Y. Hirata, T. Naito, I. Ninomiya, Heterocycles 22 (1984) 1041-1044; (d) H. Takeaki, M. Yumiko, Okiko, et al., Chem. Pharmaceu. Bull. 37 (1989) 901-906; (e) P.S. Mariano, E.W. Baxter, D. Labaree, H.L. Ammon, J. Am. Chem. Soc. 112 (1990) 7682-7692; (f) G. Varchi, A. Battaglia, C. Samorì, et al., J. Nat. Prod. 68 (2005) 1629-1631. |
| [7] |
(a) R.W. Wilkins, W.E. Judson, New Eng. J. Med. 248 (1953) 48-56; (b) R.J. Vakil, Br. Heart J. 11 (1949) 350-355; (c) M. Bleuler, W.A. Stoll, Ann. New York Acad. Sci. 61 (1955) 167-173; (d) M.F. Kossover, A.M. Goldman, CHEST 42 (1962) 170-175; (e) A.S. Leon, M.S. Belle, M. Halpern, CHEST 42 (1962) 626-634; (f) D.S. Fabricant, N.R. Farnsworth, Envir. Health Persp. 109 (2001) 69-75. |
| [8] |
X.Y. Liu, Y. Qin, Acc. Chem. Res. 52 (2019) 1877-1891. DOI:10.1021/acs.accounts.9b00246 |
| [9] |
X. Wang, D. Xia, W. Qin, et al., Chem 2 (2017) 803-816. DOI:10.1016/j.chempr.2017.04.007 |
| [10] |
R.M. Moriarty, O. Prakash, M.P. Duncan, R.K. Vaid, J. Org. Chem. 52 (1987) 150-153. DOI:10.1021/jo00377a027 |
| [11] |
O.H. Oldenziel, D. van Leusen, A.M. VanLeusen, J. Org. Chem. 42 (1977) 3114-3118. DOI:10.1021/jo00439a002 |