 2021, Vol. 32
2021, Vol. 32 


Substituted pyridines are commonly used as precursors in the synthesis of natural products [1], pharmaceuticals [2], and organocatalysts [3], as well as in ligand design [4]. However, the direct functionalization of pyridines is challenging because of their poor chemoselectivity and low reactivity [5]. Specifically, the functionalization of the C3- or C5-position is frequently ineffective without the activation of additional substituents. In addition, the modern synthetic strategies of pyridines mainly rely on transition metal catalyzed cycloaddition reactions [6]. In a complementary fashion, the direct construction of highly substituted pyridines offers an alternative strategy by utilizing abundant and inexpensive starting materials. In view of the importance of highly substituted pyridine-containing pharmaceuticals, we are interested in exploring new strategies toward the divergent synthesis of these compounds from readily available reagents, as well as investigating their applications.
Monocyclic 1,2,3-triazines are six-membered aromatic heterocycles that are electron deficient because of the three nitrogen atoms in the ring system. Their electron deficiency allows the reaction of triazines with nucleophiles [7]. Although exsiting studies are highly valuable [7-12], and have revealed efficient synthetic routes to valuable medicinal chemistry targets [7f, 7g], they typically focused on the inverse electronic Diels–Alder reaction of 1,2,3-triazines with electron-rich dienophiles [7-9] (Scheme 1a). In contrast, examples of the nucleophilic addition reactions of triazines remain rare.

|
Download:
|
| Scheme 1. Background and our proposal. | |
The divergent synthesis of various scaffolds from readily avaiable reagents are crucial to build a molecule library for drug screening and discovery. In view of the importance of pharmaceuticals, we envisioned that highly substituted pyridines c and e might be constructed via the cascade nucleophilic addition reaction of 1,2,3-triazines a with readily available acetonitrile b and ketone d derivatives, respectively (Scheme 1b). In particular, several biologically important moleculars were effectively synthesized from readily available starting materials in a few steps, obviating lengthy and laborious substrate syntheses.
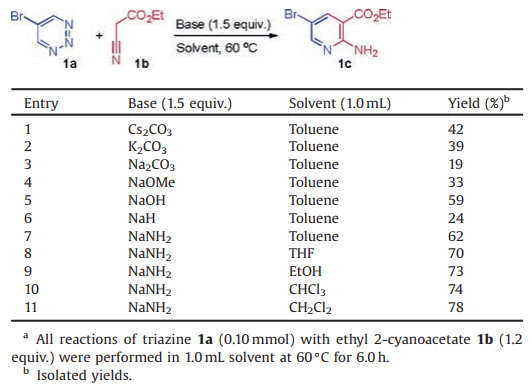
To verify the feasibility of the transformation, 5-bromo-1,2,3-triazine 1a [7b, 7g] and ethyl 2-cyanoacetate 1b were used as model substrates to commence our studies (Table 1). A range of bases were screened, and the use of NaNH2 obtained the desired 2,3,5-trisubstituted pyridine products in good yields in toluene at 60 ℃ after stirring for 6.0 h (entries 1–7). The yield was further improved by solvent screening (entries 8–11). The results indicate that dichloromethane is the best solvent (entry 11). This transformation proceeded rapidly to provide the nucleophilic addition product as a single regioisomer without the detection of any intermediates.
|
|
Table 1 Optimization of reaction condition.a |
{kind=link}
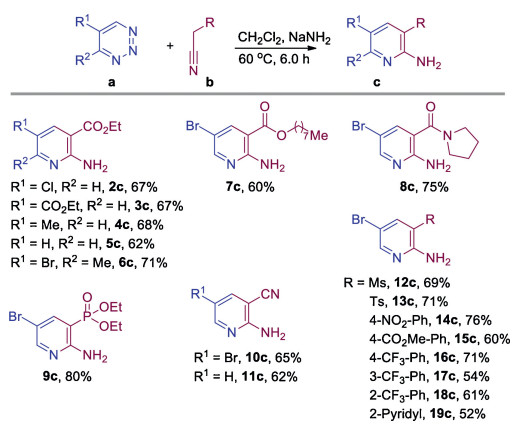
Having established the optimal reaction conditions, we initially examined the synthetic scope of various 1,2,3-triazines with ethyl 2-cyanoacetate to construct substituted pyridines (Scheme 2). Those 1,2,3-triazines bearing either a C5 substituent or without any substituents were well tolerated under our standard conditions, providing the desired products 2c–5c in good yields with no significant differences. The 4,5-disubstituted 1,2,3-triazine was effectively converted to a tetra-substituted pyridine 6c in a good yield with excellent regioselectivity. Next, acetonitriles with various substituents were used, and these reactions delivered the corresponding 2,3,5-trisubstituted pyridines 7c–19c in satisfactory yields. Among them, ranges of a-aryl substituted acetonitriles were also competent substrates, providing 3-aryl substituted pyridines 14c–19c in moderate to high yields. Varying the positions of substituents on the benzene ring, such as 4-CF3, 3-CF3 and 2-CF3 had little impact on the yields (16c–18c). The acetonitrile with an a-2-pyridyl substituent also underwent the same transformation and gave the 2,3'-bipyridine product 19c in a moderate yield.

|
Download:
|
| Scheme 2. Substrate scope. All reactions of 1,2,3-triazines a (0.20 mmol) with substituted acetonitriles b (1.2 equiv.) were performed under the standard conditions. Isolated yields. | |
{kind=link}
Having investigated the substrate scope for the reaction of 1,2,3-triazines with substituted acetonitriles, we next explored the reaction of 1,2,3-triazines with substituted ketones (detailed conditions please see Supporting information). Similarly, the screening of different bases and solvents revealed that Cs2CO3 in tetrahydrofuran (THF) provided the substituted pyridines e in excellent yields. We then investigated the scope of substrates for ketones (Scheme 3). The cyclohexyl ketone was well tolerated under the standard reaction conditions, resulting in a good yield of 2,3,5-trisubstituted pyridine 2e. Various β -aryl ketone esters were smoothly converted into the desired products 3e–8e in high yields (85%–91%). The reaction of symmetric dimethyl 3-oxopentanedioate also proceeded with 5-bromo-1,2,3-triazine, providing the desired product 9e in 84% yield. Moreover, 1,3-diphenylpropane-1,3-dione, 1-(methylsulfonyl)propan-2-one and dimethyl (2-oxopropyl)phosphonate also performed well in the current system, furnishing the desired products 10e–12e in high yields.

|
Download:
|
| Scheme 3. Substrate scope. All reactions of ketones d (0.20 mmol) with triazines a (1.5 equiv.) were performed with Cs2CO3 (1.5 equiv.) in THF at r.t. for 3.0 h. Isolated yields. | |
{kind=link}
To compare the reactivity of the substituted acetonitriles and ketones, a series of β -ketone nitriles were reacted with 5-bromo-1,2,3-triazine under the standard reaction conditions, furnishing the corresponding 3-nitrile pyridines 13e–16e in good yields rather than a-amino pyridines. In addition, various triazines could also be converted into the 3-nitriles products in high yields 17e–19e. Furthermore, this selectivity was not suppressed by varying the reaction conditions, and the results show that the ketones are more reactive than the substituted acetonitriles. Next, we investigated the preparation of bi- and terpyridines, which are known to coordinate with a wide variety of metal ions, and considerable progress has been made in their homogeneous catalysis [4]. They are also common in natural products and pharmaceuticals [1, 2]. However, traditionally, the synthesis of these compounds requires non-trivial multistep synthesis or metal-catalyzed coupling reactions. Therefore, the development of new methods to synthesize bi- and terpyridines easily is highly attractive. Interestingly, the reaction of the various 1,2,3-triazines with β -pyridine ketone ester generated the expected 2,2'-bipyridine products 20e–22e in high yields. Similarly, reactions with β -(2-methyl-pyridine) ketone and β -isoquinoline afforded the corresponding bipyridines 23e–24e in excellent yields, indicating that the current transformation is insensitive to the steric bulk of the substituents. Fortunately, the reactions of 1,2,3-triazines with diketones also afforded the 2,2': 6', 2"-terpyridine products 25e–26e in good yields, further expanding the serviceability of this transformation.
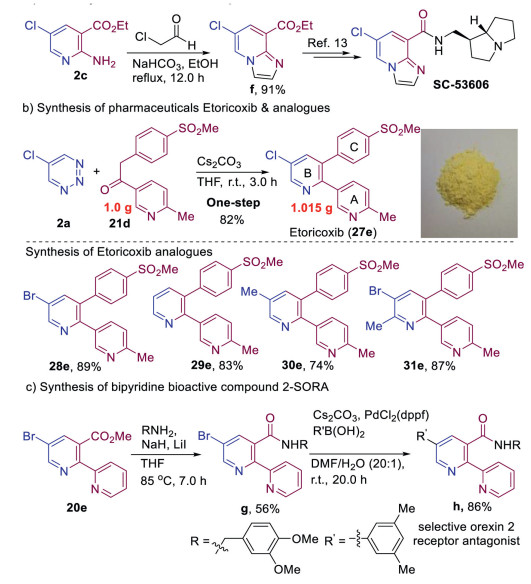
In general, the resulting substituted pyridines are convenient precursors for subsequent modification or transformation. As shown in Scheme 4, we applied this method to synthesize bioactive molecules and pharmaceuticals. Compound 2c could be converted into imidazo[1,2-a]pyridine structural unit f via a simple cyclization reaction (Scheme 4a). The f is a synthetic precursor of bioactive molecule SC-53606, which is a selective 5-HT4 receptor antagonist [13]. Moreover, we applied this reaction to the synthesis of the marketed pharmaceutical etoricoxib, which is used to treat osteoarthritis, rheumatoid arthritis, acute gouty arthritis, and ankylosing spondylitis, and can also be used to treat acute pain and chronic musculoskeletal pain [14]. The first synthesis was accomplished by Merck in a six-step sequence starting from 3-bromo-5-chloropyridin-2-ol [14a]. Fortunately, using our method, the gram-scale reaction of 5-chloro-1,2,3-triazine 2a with the corresponding ketone delivered etoricoxib 27e in 82% yield in one step (Scheme 4b). The divergent synthesis of etoricoxib with B ring modification were also expediently achieved by using various 1,2,3-triazines, these compounds (28e–31e) are attractive as potential feedstocks for the structure–activity relationship (SAR) studies of etoricoxib analogues. These transformations also proved to be effective for the synthesis of 2,3-diaryl substituted pyridines. Furthermore, to demonstrate the utility of 5-bromopyridines as effective coupling partners, we explored their application in the synthesis of bioactive bipyridine compound h, which is a selective orexin 2 receptor antagonist (2-SORA) for the treatment of insomnia [15]. Specifically, the ammonolysis reaction of the ester and Suzuki cross-coupling afforded compound h, demonstrating the tolerance of our method toward a polypyridine systems (Scheme 4c).

|
Download:
|
| Scheme 4. Synthetic applications. | |
{kind=link}
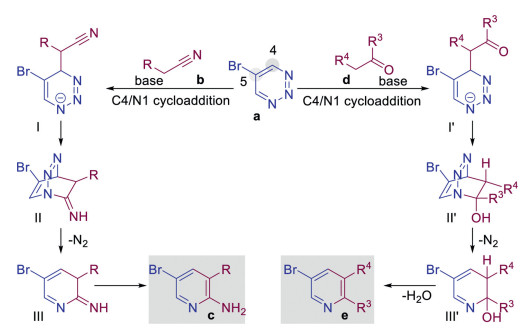
Inverse electron demand Diels–Alder reactions is a type of pericyclic reactions, in which the driving force is heat (thermal reaction) or light (photochemical reaction) [7e, 7g]. However, our reactions can also be performing at room temperature. Therefore, we consider that our reaction is more likely to be a stepwise nucleophilic addition process. Based on our experimental results, a possible mechanism is proposed in Scheme 5. Initially, in the presence of a base, the intermediate Ⅰ or Ⅰ' is formed by the addition of substituted acetonitriles or ketones to the C4-position of 1,2,3-triazines, respectively. Moreover, the intramolecular nucleophilic addition of nitrogen anion to the cyano or carbonyl group results in strained bridged-ring intermediate Ⅱ or Ⅱ', followed by the release of N2, thus forming intermediate Ⅲ or Ⅲ'. Finally, the former intermediate forms a-amino pyridines c through isomerization of the C = N double bond, whereas the latter forms pyridines e with the generation of H2O.

|
Download:
|
| Scheme 5. The proposed mechanism. | |
{kind=link}
In summary, we have developed a cascade nucleophilic addition reaction with 1,2,3-triazines and substituted acetonitriles or ketones for convenient access to a range of pyridine-containing products. Synthetically important bi- and terpyridines were also effectively obtained using this method. In particular, the marketed pharmaceutical etoricoxib, as well as its analogues were synthesized in one step with high yields. The developed method was also employed for the synthesis of several bioactive molecules. In general, this strategy is highly practical for the synthesis of pharmaceuticals and bioactive compounds. Thus, we anticipate that our methods will be widely used by the synthetic chemistry community.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (No. 21772019); Young Elite Scientist Sponsorship Program by CAST (No. 2016QNRC001); The Fundamental Research Funds for the Central Universities (No. 2019CDQYHG015); The Basic and Frontier Research Project of Chongqing (No. cstc2018jcyjAX0716); The Venture & Innovation Support Program for Chongqing Overseas Returnees (No. cx2019007). We also thank key laboratory of luminescent and real-time analytical chemistry (Southwest University), and analytical and testing center of Chongqing University.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.03.075.
| [1] |
(a) D. O'Hagan, Nat. Prod. Rep. 17 (2000) 435-446; (b) G.D. Henry, Tetrahedron 60 (2004) 6043-6061; (c) I. Nakamura, Y. Yamamoto, Chem. Rev. 104 (2004) 2127-2198; (d) M.H. Cao, N.J. Green, S.Z. Xu, Org. Biomol. Chem. 15 (2017) 3105-3129; (e) L. Li, Z. Chen, X. Zhang, Y. Jia, Chem. Rev. 118 (2018) 3752-3832. |
| [2] |
(a) W. Du, Tetrahedron 59 (2003) 8649-8687; (b) K.V. Sashidhara, R.K. Modukuri, D. Choudhary, et al., Eur. J. Med. Chem. 70 (2013) 802-810; (c) E. Vitaku, D.T. Smith, J.T. Njardarson, J. Med. Chem. 57 (2014) 10257-10274; (d) D. Verga, C.H. NâGuyen, M. Dakir, et al., J. Med. Chem. 61 (2018) 10502-10518. |
| [3] |
C.E. Müller, P.R. Schreiner, Angew. Chem. Int. Ed. 50 (2011) 6012-6042. |
| [4] |
(a) C. Kaes, A. Katz, M.W. Hosseini, Chem. Rev. 100 (2000) 3553-3590; (b) G. Chelucci, R. Thummel, Chem. Rev. 102 (2002) 3129-3170. |
| [5] |
J.A. Bull, J.J. Mousseau, G. Pelletier, A.B. Charette, Chem. Rev. 112 (2012) 2642-2713. |
| [6] |
(a) Z. Shi, T.P. Loh, Angew. Chem. Int. Ed. 52 (2013) 8584-8587; (b) J. Chen, Q. Song, C. Wang, Z. Xi, J. Am. Chem. Soc. 124 (2002) 6238-6239; (c) J.A. Varela, C. Saá, Chem. Rev. 103 (2003) 3787-3802; (d) G. Domínguez, J. Pérez-Castells, Chem. Soc. Rev. 40 (2011) 3430-3444; (e) N.S.Y. Loy, A. Singh, X. Xu, C.M. Park, Angew. Chem. Int. Ed. 52 (2013) 2212-2216; (f) A. Prechter, G. Henrion, P.F. dit Bel, F. Gagosz, Angew. Chem. Int. Ed. 53 (2014) 4959-4959. |
| [7] |
(a) A. Ohsawa, T. Kaihoh, H. Igeta, Chem. Commun. (1985) 1370-1371; (b) T. Kaihoh, T. Itoh, A. Ohsawa, et al., Chem. Phar. Bull. 35 (1987) 3952-3954; (c) T. Itoh, K. Nagata, T. Kaihoh, et al., Heterocycles 33 (1992) 631-639; (d) E.D. Anderson, A.S. Duerfeldt, K. Zhu, C.M. Glinkerman, D.L. Boger, Org. Lett. 16 (2014) 5084-5087; (e) C.M. Glinkerman, D.L. Boger, Org. Lett. 17 (2015) 4002-4005; (f) E.D. Anderson, D.L. Boger, Org. Lett. 13 (2011) 2492-2494; (g) E.D. Anderson, D.L. Boger, J. Am. Chem. Soc. 133 (2011) 12285-12292. |
| [8] |
(a) T. Sugita, J. Koyama, K. Tagahara, Y. Suzuta, Heterocycles 23 (1985) 2789-2791; (b) T. Sugita, J. Koyama, K. Tagahara, Y. Suzuta, Heterocycles 24 (1986) 29-30; (c) T. Okatani, J. Koyama, K. Tagahara, Y. Suzuta, Heterocycles 26 (1987) 595-597; (d) T. Okatani, J. Koyama, Y. Suzuta, K. Tagahara, Heterocycles 27 (1988) 2213-2217; (e) T. Okatani, J. Koyama, K. Tagahara, Heterocycles 29 (1989) 1809-1814; (f) J. Koyama, T. Ogura, K. Tagahara, Heterocycles 38 (1994) 1595-1600; (g) A. Díaz-Oritz, A. de la Hoz, P. Prieto, et al., Synlett 2 (2001) 236-237. |
| [9] |
(a) T. Itoh, A. Ohsawa, M. Okada, T. Kaihoh, H. Igeta, Chem. Pharm. Bull. 33 (1985) 3050-3052; (b) T. Itoh, M. Okada, K. Nagata, K. Yamaguchi, A. Ohsawa, Chem. Pharm. Bull. 38 (1990) 2108-2111. |
| [10] |
(a) A. Ohsawa, H. Arai, H. Ohnishi, H. Igeta, Chem. Commun. (1980) 1182-1183; (b) A. Ohsawa, H. Arai, H. Ohnishi, H. Igeta, Chem. Commun. (1981) 1174-1174; (c) A. Ohsawa, H. Arai, H. Ohnishi, et al., J. Org. Chem. 50 (1985) 5520-5523; (d) A. Ohsawa, T. Kaihoh, T. Itoh, et al., Chem. Pharm. Bull. 36 (1988) 3838-3848. |
| [11] |
(a) H. Neunhoeffer, M. Clausen, H.D. Vöetter, et al., Liebigs Ann. Chem. (1985) 1732-1751; (b) H. Neunhoeffer, R. Bopp, W. Diehl, Liebigs Ann. Chem. (1993) 367-373; (c) M. M. Mättner, H. Neunhoeffer, Synthesis (2003) 413-425. |
| [12] |
(a) A.S. Duerfeldt, D.L. Boger, J. Am. Chem. Soc. 136 (2014) 2119-2125; (b) C.M. Glinkerman, D.L. Boger, J. Am. Chem. Soc. 138 (2016) 12408-12413; (c) J. Zhang, V. Shukla, D.L. Boger, J. Org. Chem. 84 (2019) 9397-9445. |
| [13] |
D.P. Becker, D.L. Flynn, A.E. Moormann, et al., J. Med. Chem. 49 (2006) 1125-1139. |
| [14] |
(a) D.Dubé, R.Fortin, R.Friesen, J.Y.Gauthier, Z. Wang, WO9803484A1, (1998); (b) R.W. Friesen, C. Brideau, C.C. Chan, et al., Bioorg. Med. Chem. Lett. 8 (1998) 2777-2782; (c) I.W. Davies, J.F. Marcoux, E.G. Corley, et al., J. Org. Chem. 65 (2000) 8415-8420; (d) E. Zhang, J. Tang, S. Li, et al., Chem. Eur. J. 22 (2016) 5692-5697. |
| [15] |
(a) S.P. Mercer, A.J. Roecker, S. Garson, et al., Bioorg. Med. Chem. Lett. 23 (2013) 6620-6624; (b) A.J. Roecker, S.P. Mercer, J.D. Schreier, et al., ChemMedChem 9 (2014) 311-322. |