 2021, Vol. 32
2021, Vol. 32 


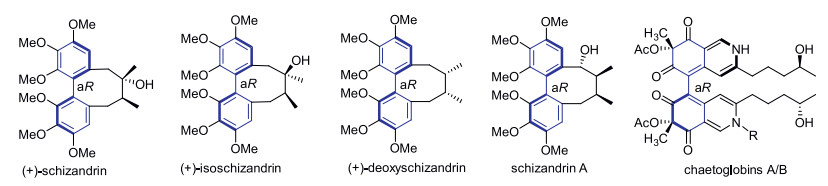
Axially chiral diphenyl motif extensively exists in a number of bioactive natural and unnatural compounds [1]. Compared to many other biaryls (such as BINOLs) and sp3 carbon-based chiral centers, building axial chirality of diphenyls is still full of challenges [1a, 1c]. For limited availability of practically useful scale-up methods, enantioselective synthesis of the natural products with an axial diphenyl core remains as a very difficult mission yet [2]. For example, chaetoglobins A/B are two characteristic diphenyl natural products having high substituted atropisomerism [3a] (Fig. 1). Recently, Kozlowski's group successfully established axial chirality of chaetoglobin A using a vanadium-catalyzed oxidative homo-phenol coupling (dimerization) in a relatively low yield, and the atropoisomers were uneasy to be completely separated [4]. Schizandrin family is another type of diphenyl natural products [3b]. Lin's group once developed a nickel-catalyzed asymmetric Ullmann coupling to construct axial chirality of (+)-isoschizandrin, in which recrystallization was needed to increase the ee value of product [5]. Even so, asymmetric synthesis of deoxyschizandrin has not been achieved yet [6]. As the crucial step to construct the axial chirality in the synthesis of such diphenyl compounds, the currently available methodologies are of urgency for further improvement, including proper scalable oxidative phenol-phenol couplings for generating diphenyl products of high enantiopurity.

|
Download:
|
| Fig. 1. Several natural products containing an axially chiral diphenyl core. | |
A considerable strategy to establish axially chiral diphenyls is intramolecular coupling of two phenols linked with a chiral tether, which is not only expected to deliver satisfying atropisomeric ratio during the C(sp2)-C(sp2) bond formation, but also suitable for the synthesis of both homo- and hetero-coupled diphenols [1a, 1c] (Fig. 2). Among commonly available chiral materials, the C2-symmetrical tartaric acid and its derivatives are economic choices for such chirality transfer in the above mentioned dehydrogenative phenol-phenol coupling. Lipshutz and coworkers once attempted to utilize tartaric acid derivative and cyanocuprate to synthesize the chiral axis and achieved a single isomer [7]. Wynberg and Feringa also developed a copper/chiral amine oxidation system for the preparation of axial binaphthols [8] and a variety of copper/amine combinations have also been reported in the following years [9]. In 2011, Yamada and coworkers explored the synthesis of chiral axial diphenols by intramolecular oxidative homo-coupling with a combination of CuCl2 and n-BuNH2 [10a], and later applied to the synthesis of highly oxygenated diaryl ethers featured in ellagitannins [10b]. Unfortunately, dehydrogenative hetero-coupling between two different phenols still keeps unsolved yet.

|
Download:
|
| Fig. 2. Illustration of a protocol for the chiral-axis formation of deoxyschizandrin by an intramolecular dehydrogenative coupling of phenols. | |
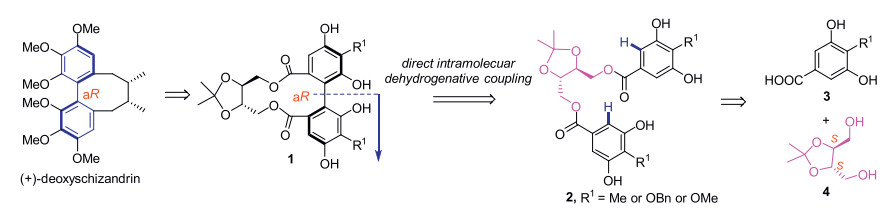
Inspired by the above pioneer works, we herein wish to report a practically scalable procedure of intramolecular dehydrogenative coupling to prepare gram-scale quantity of enantiopure axially chiral diphenyls by either homo- or hetero-coupling of two electron-rich phenols. This new method is applicable in total synthesis of the above mentioned natural products having a chiral diphenyl axis, such as (+)-deoxyschizandrin (Fig. 2). As shown in Fig. 2, the C2-symmetrical tartaric acid-derived diol 4 would be introduced by esterification with two benzoic acids 3 to furnish bis-benzoate substrates 2. The expected product, axially chiral diphenyl 1, is a useful common synthon for the synthesis of chaetoglobins A and B, and deoxyschizandrin as well. We propose that the dioxolane motif of the chiral tether would help to fix the gauche conformation of the diol 4 and thus facilitate the crucial dehydrogenative coupling [11]. It is noteworthy that the C2 symmetric diol 4 is capable to link two different benzoic acids through a two-step esterification procedure, providing us more flexibility to examine a wide range of diphenol substrates including those for hetero-coupling.
The reaction was initially performed in MeOH with CuCl2 (1 equiv.) and amine (4 equiv.) at room temperature for 30 min under nitrogen atmosphere, and the substrate (1 equiv.) concentration was set at 0.04 mol/L. Our study showed that a higher loading of CuCl2 (3.5 equiv.) was needed to complete the reaction [8, 9, 12]. The released halide ion was suspected to deactivate the oxidative potential of certain unidentified Cu(II) complexes in the reaction [13]. Using compound 5a as the substrate, effect of the amine ligand was examined upon the diastereomeric ratio of products (Table 1). Two cyclic diphenols, (aR)-6a (major product) and (aS)-6a′ (minor product), were isolated in a combined yield of 78% using n-propylamine or n-butylamine, respectively (Table 1, entries 1 and 2, accordingly with 17% and 35% de). While, the corresponding reactions with t-butylamine or triethylamine did not happen (entries 3 and 4). They might mention that steric hindrance of the amines is unfavorable to formreactive oxidative complexes of Cu(II) [9]. A weaker base, aniline, could not promote this reaction either (entry 6). The de value of the products increased up to 55% when cyclohexylamine was applied as the ligand, though no significant improvement was found of the yield (76%, entry 5). When benzylamine was used, the de value was further improved up to 75% (entry 7). However, electronic effect was moderate when the phenyl groups of the benzylamines were differently substituted. For example, the product de values from the reactions with p-methoxyphenylmethylamine and p-fluorophenylmethylamine reduced to 60% and 40%, respectively (entries 8,9). However, stereochemical effect of the benzylamine ligand is significant on the ratio of two axial products. The de value increased up to 89% when (R)-α-methyl-benzylamine was used, delivering 6a and 6a′ (ratio 94.5:5.5) in 80% combined yield (entry 10). While, (S)-α-methyl-benzylamine was proved to be poorly matched to the corresponding reaction, affording 6a and 6a′ with 67% de (dr 83.5:16.5) and a lower yield (entry 11). The diamine ligands (ethylenediamine, or trans-1,2-diaminocyclohexane) totally inhibited the reaction (entries 12,13). Therefore, the optimal reaction conditions for this intramolecular dehydrogenative coupling were determined as follows: diphenol substrate 5a (0.4 mmol), CuCl2 (1.4 mmol), (R)-α-methyl-benzylamine (5.6 mmol) in MeOH (10 mL) under nitrogen atmosphereat room temperature for 30 min.
|
|
Table 1 Screening of the amine ligands in the intramolecular dehydrogenative homo-coupling of diphenol.a |

The absolute configuration of the minor product (aS)-6a′was confirmed by a single crystal X-ray diffraction experiment (Fig. 3). In addition, the CD spectrum (MeOH) of (aS)-6a′ shows a positive Cotton effect at λ 232 nm (Δε 42.3), λ 267 nm (Δε 7.5), while the major product (aR)-6a shows a negative Cotton effect at λ 232 nm (Δε -43.1), λ 267 nm (Δε -18.4) (Fig. 3). Such CD characterizations were generally observed in all pairs of these products (for more details, see Supporting information), and thus are helpful for quick elucidation of the absolute stereochemistry of chiral axis of chiral axial diphenols.

|
Download:
|
| Fig. 3. Determination of the absolute stereochemistry of the major product 6a (CD/green line) and minor product 6a′ (X-ray, and CD/purple line). | |
The substrate scope was then investigated using the optimized combination of CuCl2 and (R)-α-methyl-benzylamine (Table 2). In the first group, homo-couplings of various resorcinols having a -Me, -OMe, or -OBu group at the meta position could be smoothly carried out, affording expected products in good yields, but in variable de values (6a/6a′ (89%), 6b/6b′ (33%), 6c/6c′ (67%) and 6d/6d′(87%), respectively). The bulky substituents at the meta position seemed to favor the formation of the major diphenyl products by "buttressing" the ortho substituents, and thus prevent their rotation in the transition state [1a, 1c, 14]. While, those highly reducible phenols resulted in a complicated mess situation, and no products were detected in these reactions (Table 2, 6e, 6f and 6g). In the meanwhile, a phenol substrate of lower reducibility did not give reaction either (Table 2, 6h). These observed results strongly mentioned us that dihydroxybenzoates are well matched the oxidizing potency of the combination of CuCl2 and (R)-α-methylbenzylamine in MeOH.
|
|
Table 2 Substrate-scope examination.a, b, c |

{kind=link}
{kind=link}
{kind=link}
The second group of substrates was specially designed for hetero-couplings, among which a variety of different resorcinol pairs were applied as coupling partners. All the examined resorcinols were well done under standard reaction conditions, providing the corresponding hetero-coupled axial diphenols. Compared to those of 6i (R1 = H, R1′ = OMe, 33% de) and 6j (R1 = H, R1′ = OBn, 20% de), the de value of 6m (R1 = Me, R1′ = H, 83% de) is much higher. It mentions that -Me (R1) may have a larger van der Waals radii compared to that of -OMe and -OBn, which is beneficial to "buttressing" the ortho substituents and makes the transition state configuration more stable [14a]. Satisfactory results were also observed in the cases of 6k (R1 = Me, R1′ = OBn, 91% de), 6l (R1 = Me, R1′ = OMe, 82% de). Again, syntheses of 6o, 6p, and 6q unfortunately failed, due to their unmatched redox potencies. The atropisomers of 6a/6a′, 6d/6d′, 6k/6k′, and 6m/6m′ could be separated directly by silica gel column chromatography. Other pairs of atropisomers could be separated by silica gel column chromatography after phenolic O-allylation of the oxidative products. Scalability of the oxidative coupling system with CuCl2/(R)-α-methylbenzylamine was also examined. The reaction of 5a (6.8 g, 15 mmol scale) was carried out smoothly in the laboratory, affording 6a (5.2 g, 74% yield) and 6a′ (301 mg, 4% yield) with similar standard of de value and isolated yield as that of milligram scale.
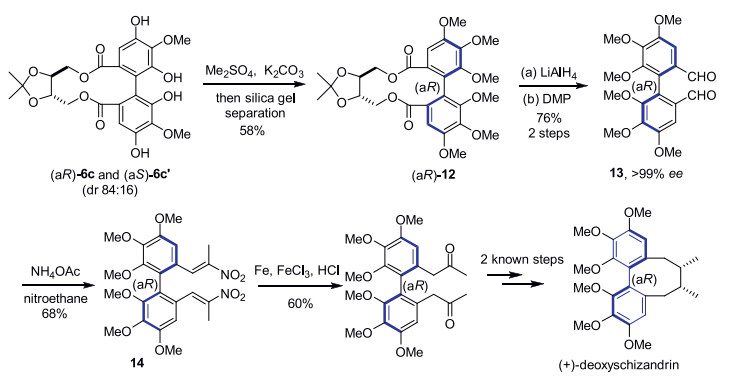
Transformations of representative product 6c were explored for the formal total synthesis of (+)-deoxyschizandrin (Scheme 1). At first, enantiopure axially chiral biphenol (aR)-12 was prepared from a diasteromeric mixture of 6c and 6c′ after phenolic O-methylation in 58% yield (Note: the two diastereomers were separated by silica gel column chromatography). Removal of the chiral auxiliary was accomplished with LiAlH4 reduction and Dess–Martin oxidation, furnishing benzaldehyde 13 (76% yield, 2 steps from 12). Chiral HPLC analysis showed that ee value of 13 was greater than 99% [15]. Treatment of aldehyde 13 with ammonium acetate and nitroethane afforded compound 14 in 68% yield. Reduction of both nitroolefin functionalities of 14 with Fe/FeCl3/HCl provided the known ketone intermediate 15 (60% yield) to (+)-deoxyschizandrin [16].

|
Download:
|
| Scheme 1. A formal total synthesis of (+)-deoxyschizandrin. | |
{kind=link}
In summary, we have successfully developed a scalable intramolecular dehydrogenative phenol-phenol coupling under mild conditions with CuCl2 and (R)-α-methylbenzylamine, and a series of enantiopure axially chiral biphenols have been synthesized accordingly. The newly developed method, aided by a removable tether of L-(+)-tartaric acid-derived diol, is eligible for both the homo- and hetero-couplings of two resorcinol units. This work greatly expands the substrate range and application in organic synthesis of chiral axial compounds. We believe that the above new scalable dehydrogenative coupling reaction and the corresponding enantiopure biphenyl products will be useful in the related study of natural and unnatural axial compounds and materials.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support by National Key Research and Development Program of China (No. 2018YFC0310900), the National Natural Science Foundation of China (Nos. 21472087, 21532002, 21901112, 21778031, 21761142001), and Jiangsu Provincial Department of Science and Technology (No. BK20190277) is greatly appreciated. The authors also thank Drs. Sheng Wang and Fei Lv for their participating the early-stage study of this work.
| [1] |
(a) G. Bringmann, A.J.P. Mortimer, P.A. Keller, et al., Angew. Chem. Int. Ed. 44 (2005) 5384-5427; (b) G. Bringmann, M.C. Günther, O. Schupp, S. Taster, Biaryls innature: a multifacettedclass of stereochemically, biosynthetically, and pharmacologically intriguing secondary metabolites, in: W. Herz, H. Falk, G.W. Kirby, R.E. Moore (Eds.), Progress in the Chemistry of Organic Natural Products, 82, Springer, New York, 2001, pp. 1-293; (c) G. Bringmann, T. Gulder, T.A.M. Gulder, M. Breuning, Chem. Rev. 111 (2011) 563-639; (d) F. Lv, Z.J. Yao, Sci. China Chem. 60 (2017) 701-720. |
| [2] |
(a) T. Qin, S.L. Skraba-Joiner, Z.G. Khalil, et al., Nat. Chem. 7 (2015) 234-240; (b) B.B. Liau, B.C. Milgram, M.D. Shair, J. Am. Chem. Soc. 134 (2012) 16765-16772. |
| [3] |
(a) J.M. Gao, S.X. Yang, J.C. Qin, Chem. Rev. 113 (2013) 4755-4811; (b) M.C. Kozlowski, B.J. Morgan, E.C. Linton, Chem. Soc. Rev. 38 (2009) 3193-3207. |
| [4] |
H. Kang, C. Torruellas, J. Liu, M.C. Kozlowski, Org. Lett. 20 (2018) 5554-5558. DOI:10.1021/acs.orglett.8b02183 |
| [5] |
W.W. Chen, Q. Zhao, M.H. Xu, G.Q. Lin, Org. Lett. 12 (2010) 1072-1075. DOI:10.1021/ol1000632 |
| [6] |
(a) T. Biftu, B.G. Hazra, R. Stevenson, J. Chem. Soc. Perkin Trans. I 1 (1979) 2276-2281; (b) T. Takeya, T. Okubo, S. Nishida, S. Tobinaga, Chem. Pharm. Bull. 33 (1985) 3599-3607. |
| [7] |
B.H. Lipshutz, F. Kayser, Z.P. Liu, Angew. Chem. Int. Ed. 33 (1994) 1842-1844. DOI:10.1002/anie.199418421 |
| [8] |
B. Feringa, H. Wynberg, Bioorg. Chem. 7 (1978) 397-408. DOI:10.1016/0045-2068(78)90031-7 |
| [9] |
(a) J. Brussee, J.L.G. Groenendijk, J.M. teKoppele, A.C.A. Jansen, Tetrahedron 41 (1985) 3313-3319; (b) M. Smrčina, M. Lorenc, V. Hanuš, P. Sedmera, P. Kočovský, J. Org. Chem. 579 (1992) 1917-1920; (c) Z. Xu, M.C. Kozlowski, J. Org. Chem. 67 (2002) 3072-3078; (d) X.L. Li, J. Yang, M.C. Kozlowski, Org. Lett. 3 (2001) 1137-1140; (e) P. Miko, J. Roithová, D. Schröder, J. Lemaire, H. Schwarz, M.C. Holthausen, Chem. Eur. J. 14 (2008) 4318-4327; (f) M. Nakajima, I. Miyashi, K. Kanayama, S. Hashimoto, J. Org. Chem. 64 (1999) 2264-2271. |
| [10] |
(a) N. Asakura, S. Fujimoto, N. Michihata, et al., J. Org. Chem. 76 (2011) 9711-9719; (b) T. Hirokane, Y. Hirata, T. Ishimoto, K. Nishii, H. Yamada, Nat. Commun. 5 (2014) 3478. |
| [11] |
(a) B.H. Lipshutz, B. James, S. Vance, I. Carrico, Tetrahedron Lett. 38 (1997) 753-756; (b) B.H. Lipshutz, Y.J. Shin, Tetrahedron Lett. 39 (1998) 7010-7020. |
| [12] |
(a) Y. Kasai, N. Michihata, H. Nishimura, T. Hirokane, H. Yamada, Angew. Chem. Int. Ed. 51 (2012) 8026-8029; (b) N. Michihata, Y. Kaneko, Y. Kasai, et al., J. Org. Chem. 78 (2013) 4319-4328; (c) H. Takeuchi, K. Mishiro, Y. Ueda, et al., Angew. Chem. Int. Ed. 54 (2015) 6177-6180; (d) B. Fering, H. Wynberg, Bioorg. Chem. 7 (1978) 397-408. |
| [13] |
A.S. Hay, H.S. Blanchard, G.F. Endres, J.W. Eustance, J. Am. Chem. Soc. 81 (1959) 6335-6336. DOI:10.1021/ja01532a062 |
| [14] |
(a) G. Bott, L.D. Field, S. Sternhell, J. Am. Chem. Soc. 102 (1980) 5618-5626; (b) M. Rieger, F.H. Westheimer, J. Am. Chem. Soc. 72 (1950) 19-28. |
| [15] |
A.I. Meyers, T.D. Nelson, H. Moorlag, D.J. Rawson, A. Meier, Tetrahedron 60 (2004) 4459-4473. DOI:10.1016/j.tet.2004.01.095 |
| [16] |
A.R. Carroll, R.W. Read, W.C. Taylor, Aust. J. Chem. 47 (1994) 1579-1589. DOI:10.1071/CH9941579 |