 2021, Vol. 32
2021, Vol. 32 


 Prof. Hujun Xie received his Bachelor's degree (2004) form Zhejiang University, and obtained Ph.D. degree from Xiamen University under the supervision of Prof. Zexing Cao. From 2010 to 2014, as a post doctor, he studied at the Hong Kong University of Science and Technology under the supervision of Prof. Zhenyang Lin and Zhejiang University under the supervision of Prof. Wenjun Fang. From 2017 to 2018, as a visiting scholar, he studied at the Fudan University under the supervision of Prof. Xin Xu. He joined Zhejiang Gongshang University in 2009, and became a full professor (2018) in the Department of Applied Chemistry. He is a top talent of West Lake Scholars in Zhejiang Gongshang University. His research focuses on the DFT calculations for homogeneous catalysis, especially in the field of nickel- and palladium-catalyzed cross-coupling reactions.
Prof. Hujun Xie received his Bachelor's degree (2004) form Zhejiang University, and obtained Ph.D. degree from Xiamen University under the supervision of Prof. Zexing Cao. From 2010 to 2014, as a post doctor, he studied at the Hong Kong University of Science and Technology under the supervision of Prof. Zhenyang Lin and Zhejiang University under the supervision of Prof. Wenjun Fang. From 2017 to 2018, as a visiting scholar, he studied at the Fudan University under the supervision of Prof. Xin Xu. He joined Zhejiang Gongshang University in 2009, and became a full professor (2018) in the Department of Applied Chemistry. He is a top talent of West Lake Scholars in Zhejiang Gongshang University. His research focuses on the DFT calculations for homogeneous catalysis, especially in the field of nickel- and palladium-catalyzed cross-coupling reactions. |
With quick development of hardware, software, and computational methodologies, theoretical calculations have become a very useful tool to investigate mechanistic details and selectivity of transition-metal-catalyzed reactions [1-5]. Calculations can obtain potential energy surfaces and thus offer extra supplement for experiments [6, 7]. Over the past decades, theoretical calculations have made great contributions to explore the origins for ligand-mediated selectivity in nickel- and palladium-catalyzed coupling reactions [8-12].
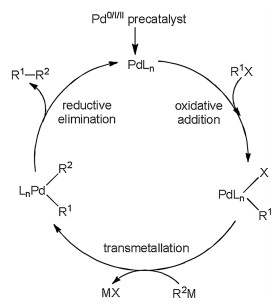
Great progresses have been obtained to comprehend important steps in coupling reactions [13-17], and the mechanism generally includes several catalytic steps, thus it is hard to explore the mechanism only from experiments. With the contributions from calculations, it is well known that Pd-catalyzed reaction mechanism commonly involves a Pd0/Pd2+ catalytic cycle as shown in Fig. 1. It mainly includes oxidative addition, transmetallation and reductive elimination to yield final product, accompanied by the regeneration of palladium catalyst. Ni-catalyzed reactions generally have similar mechanism.

|
Download:
|
| Fig. 1. Possible reaction mechanisms. | |
{kind=link}
To explain reaction selectivity in coupling reactions, experimental studies on mechanisms are limited because transition state can not be found, which is an important factor to explore the reaction selectivity, thus calculations help to supply valuable insights. Computing detailed potential energy surfaces (PESs) of full catalytic cycle is an arduous task, while it provides new insights into the barrier for each step [18, 19]. Calculations elucidate the selectivity through considering various catalytic pathways to give different products, and assist to design new catalysts to control reaction selectivity of coupling reactions [20, 21].
For coupling reactions, selectivity is important because side reactions including β-hydride elimination or isomerization exist. Selectivity is often controlled by transition metals themselves or ligands around them, and the nature of ligand is seemed as decisive variable, since the variation of ligands is more diverse than transition metals. Thus more attentions have been paid to explore possible effects of ligand on each step of catalytic cycle. In this review, we focused on the selectivity in nickel- and palladium-catalyzed coupling reactions. Previous calculations have obtained great achievements to explore the mechanisms of coupling reactions [22-25]. Catalytic mechanisms were studied step by step to explore rate- and selectivity-determining step. As we know, theoretical studies on ligand-controlled selectivity in these reactions have not been widely reviewed up to now [26]. This review focuses on theoretical studies of ligand-controlled selectivity in Pd- and Ni-catalyzed coupling reactions, and divides into four sections, introduction, computational methods, selectivity control by ligands in Ni- and Pd-catalyzed coupling reactions as well as summary and future perspectives. It highlights ligand factors to control product selectivities in coupling reactions. They contain steric repulsions between ligands and substrate, electronic effects determined via substrate-metal interactions and dispersion effects. This review outlines our work and some significant contributions from other groups on ligand-controlled selectivity in coupling reactions.
2. Computational methodsWith quick development of theoretical methods and computer power, research systems have dramatically expanded and many important issues can be solved computationally. In order to compare theoretical and experimental results, the selected computational method should be accurate enough, which needs to consider different factors including dispersion [27-38], solvation [39-41] and vibrational corrections [42].
Currently, B3LYP functional is still widely applied for conformation optimization of Pd and Ni species [43-47], while some faults of this functional have been revealed [48-53]. The major problems involve unreasonable description for dispersion interaction, which is very important for Ni- and Pd-catalyzed reactions with large systems. One method to solve this issue is to develop new functions parametrized with dispersion, and the other method is related to add a semiclassical dispersion correction into the functions. The M06 series developed by Truhlar group [54-58] and the density functional method containing dispersion (DFT-D3) developed by Grimme [59-60], [61] are two typical examples to solve this issue. These two functionals are becoming increasingly popular in nickel- and palladium-catalyzed coupling reactions, because these computational methods could provide good description of potential energy surface for catalytic cycle.
Steric and electronic effects of ligands are very important to design reasonable ligands in coupling reactions. Previous viewpoint generally considers that steric effect is repulsive, and recent research counterintuitively proposed that large hydrocarbon compounds are stabilized more strongly than small molecules due to dispersion interactions. This attractive dispersion force is very important in coupling reactions. The work done by Schoenebeck et al. [62] confirmed the dispersion role on the structures and reactivities of PtBu3 (cone angle of 182°) and P(iPr)(tBu2) (cone angle of 175°) ligands in palladium-catalyzed reactions.
For the reaction of 4-chlorophenyl triflate with palladium catalyst, the transition states of bisphosphine ligand involving aromatic C-O bond oxidative addition has been found by DFT calculations with dispersion correction, while the same method without dispersion correction leads to loss of a phosphine ligand, indicating that dispersion is very important for large phosphine ligand. For small ligand P(iPr)(tBu2), the C-Cl oxidative addition is preferred for PdL and the C-OTf oxidative addition is preferred for PdL2. Further calculations showed that different selectivity can be achieved depending on whether dispersion interaction was considered or not. The other well-established parameters including percent buried volume (%Vbur) [63] and Tolman cone angle θ [64-70] to consider electronic and steric effects of ligands were also used to design ligands.
3. Selectivity control by ligands in Ni- and Pd-catalyzed coupling reactions 3.1. Evolution of ligand design of Pd and Ni catalystsOver the past decade, the profound effect of ligand selections on the coupling reactions by Pd and Ni catalysts were explored. Many ligands including monodentate (phosphine ligand) and bidentate (P-N, P-P, P-O) ligands have been developed, and ligand parameters including percent buried volume, concept of bite angle, as well as cone angle were also introduced [64-72].
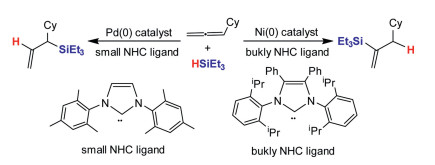
Previouslly, small triarylphosphine ligands were widely selected in Ni- and Pd-catalyzed coupling reactions, while significant contributions from Buchwald group elucidated that bulky dialkylbiarylphosphine ligands expand the scope and efficiency of catalytic systems [64-70]. Pd(Ⅱ) catalysts possessing phosphine ligands displayed good catalytic efficiency with outstanding stereo- and regio-selectivities [71-72]. Fu and coworkers [73] investigated the palladium-catalyzed Suzuki–Miyaura reactions of C(sp2)-triflate and C(sp2)-halide with aryl boronic acid by using PtBu3 and PCy3 ligands, and the research clarified that the chemoselectivity can be controlled via selecting suitable ligand, leading to a reversal selectivity for the oxidative addition of Ar-Cl versus Ar-OTf. The N-heterocyclic carbenes (NHC) are new class of ligands that grow quickly in Pd- and Ni-catalyzed coupling reactions. The steric effect and strong σ-donating ability of NHC ligands can activate and stabilize Pd and Ni centers in catalytic reactions [74, 75]. Montgomery and coworkers [76] studied the hydrosilylation of allene catalyzed by Pd and Ni complexes with NHC ligands. It has been found that the reaction regioselectivity can be controlled by selecting different metals and NHC ligands. Bulky Ni-NHC catalyst yields alkenylsilanes, and small Pd-NHC catalyst produces allylsilanes.
The selectivity in Pd- and Ni-catalyzed reactions can be achieved via mediating the electronic and steric effects of ligands [77-79], and appropriate ligand is often picked out by many screening experiments. With rapid developments of theoretical methods, software and computing power, the selection of calculated methods for reasonable design of ligands has attracted extensive interest [80-82]. It is impossible that one ligand can be used for all Pd- and Ni-catalyzed cross-coupling reactions, but instead, suitable ligand is selected for a particular reaction [83].
3.2. Ni catalysts 3.2.1. C-O bonds activationThe development of ligand-controlled methods for reaction selectivity remains a critical challenge in Pd- and Ni-catalyzed coupling reactions [84-87]. Recent researches found that Ni catalysts are very effective for C-O bonds activation, including carbamates, esters, sulfamates and aryl alkoxides, and many computations were performed to understand the mechanism for selectivity [88-91]. Most of catalytic cycles in coupling reactions is involved in the Ni0/Ni2+ path, while the odd oxidation states containing the Ni1+/Ni3+ path are also possible [92]. Herein, we introduced new progresses on theoretical calculations on the reaction mechanisms and origins for ligand-controlled selectivity.
Hong and Houk [93] reported the reaction mechanisms in nickel-catalyzed activation of aryl esters by phosphine ligands, and the switchable selectivity for C(acyl)-O and C(aryl)-O bonds cleavage of aryl esters were considered by DFT computations (Scheme 1). The calculations demonstrated that the Ni catalyst with bidentate dcype ligand can cleave C(acyl)-O bond by three-membered transition state (TS). The C(acyl)-O bond with low bond dissociation energy leads to low distortion energy of TS, facilitating the activation of C(acyl)-O bond. For monodentate PCy3 ligand, a five-centered TS for C(aryl)-O bond cleavage was found. The extra Ni -O interaction energy may override the distortion penalty and facilitates C(aryl)-O bond cleavage.

|
Download:
|
| Scheme 1. Nickel-catalyzed C(aryl)-O and C(acyl)-O bonds cleavage of aryl ester by different ligands. | |
{kind=link}
Ni catalysts with different ligands can control reaction stereoselectivity for Suzuki-Miyaura coupling reactions of benzylic pivalates derivatives and boronates [94-96]. Tricyclohexylphosphine (PCy3) ligand yields the reserved stereochemistry at the carbon center (Scheme 2), and SIMes ligand (1, 3-dimesityl-4, 5-dihydroimidazol-2-ylidene) generates the stereocenter inverted product. Our group [94] and Hong [95] performed the calculations to study the reaction mechanism and stereoselectivity through ligand control. Oxidative addition (OA) step decides the stereoselectivity involving two different TSs, and concerted OA by a cyclic TS forms stereoretention, and a TS like SN2 back side attack can invert the stereocenter of carbon atom. The obvious difference between two TSs is related to angle distortion of ligand-nickel-substrate, and the ligand can control reaction stereoselectivity by distinguishing different angle distortion. For PCy3 ligand, the ligand-Ni interaction is dominated by σ-donation and it shows slight angle distortion. Easy angle distortion by PCy3 ligand prefers a cyclic TS for OA and can retain stereo center. For SIMes ligand, the extra d-p back-donation from Ni to carbene can improve the rigidity of ligand-Ni bond, and angle distortion becomes difficult. Percent buried volume models and distortion/interaction energy analysis indicated that the interactions between organic part and catalyst mediate the stereoselectivity for Ni-SIMes and Ni-PMe3 catalysts.

|
Download:
|
| Scheme 2. The stereoselectivity is controlled by different ligands in Ni-catalyzed reaction of benzylic pivalates derivatives with boronates. | |
{kind=link}
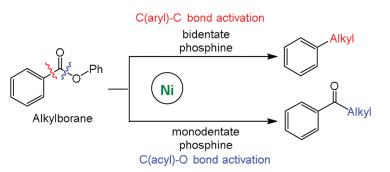
Rueping et al. reported ligand-controlled selectivity in Ni-catalyzed Suzuki-Miyaura reactions of alkyl organoboron reagents and aromatic esters [97], and the calculations indicated that oxidative addition determines reaction selectivity (Fig. 2). Ni catalysts with monodentate phosphin ligands (PBu3 and PCy3) facilitate the O-C(acyl) bond cleavage to give ketone product, while nickel catalyst carrying bidentate ligands (dcype) facilitate the C-C(aryl) bond activation to give alkylated product through decarbonylation step. The barrier for O-C(phenyl) bond oxidative addition is much higher than that of O-C(acyl) and C-C(aryl) bonds, and selectivity switch is controlled by different phosphine ligands. The increased steric repulsion by PBu3 ligand can destabilize the transition state for C-C(aryl) activation, and facilitates the O-C(acyl) bond activation (Fig. 3). For the TS of C-C(aryl) bond activation, the geometry can maximize the interaction between alkyl P-substituents and substrate (Fig. 3). In contrast, for the TS of O-C(acyl) bond activation, the geometry can minimize the interaction between alkyl P-substituents and substrate.

|
Download:
|
| Fig. 2. The selectivity in nickel-catalyzed reactions of alkyl organoboron reagents and aromatic esters. | |
{kind=link}

|
Download:
|
| Fig. 3. Transition states for the O-C(acyl) and C-C(aryl) bonds activation by Ni(PnBu3)2 and Ni(dcype) catalysts. | |
{kind=link}
The aldehydes and alkynes reductive coupling reactions by nickel catalysts have attracted wide attentions in experiments, and the mechanisms and origins for stereo- and regio-selectivities of these reactions have been explored via the DFT calculations [98-101].
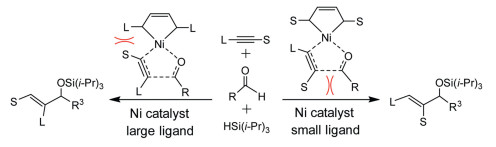
Liu and Houk [102] reported the regioselectivity in Ni-catalyzed alkyne and aldehyde reductive coupling reaction using silane as reducing agents (Scheme 3). The selectivity is originated from the transition state of oxidative addition that can be mediated via steric repulsions between hindered NHC ligand and the substituents of alkyne.

|
Download:
|
| Scheme 3. The regioselectivity in Ni-catalyzed alkyne and aldehyde reaction. | |
{kind=link}
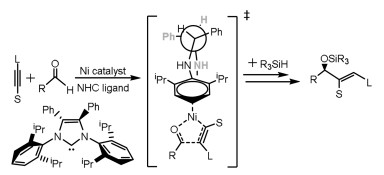
The NHC ligands not only control the regioselectivity for the reactions of aldehydes and alkynes, but also control their enantioselectivities. Liu and Montgomery reported that the NHC ligands can simultaneously control regio- and enantio-selectivities (Scheme 4) [103]. Computational studies showed that the origin of selectivity is ascribed to steric repulsions between aldehydes and NHC ligand.

|
Download:
|
| Scheme 4. Ni-catalyzed reactions of aldehydes and alkynes. | |
{kind=link}
3.3. Pd catalysts 3.3.1. Activation of aryl-halides, alkyl-halides and C-OTf bonds
The calculations were performed to investigate the origins of selectivity in palladium catalysis [104-106]. The manifold of oxidative addition step for the reaction of phenyl chloride and phenyl bromide by palladium catalyst has been studied, and the dispersion-corrected density functional theory (B3LYP-D2) was selected to evaluate substrate, method and ligand effects on computational results [107-109]. It has been demonstrated that oxidative addition by bisphosphine ligand is facile for small PPh3 and PCy3 ligands, while electron-rich and bulky ligands SPhos and PtBu3 facilitate the low-coordinate complexes.
The regio- and stereo-selectivities for Suzuki-Miyaura reactions have attracted extensive interest [110, 111]. Maseras et al. investigated the regioselectivity in Pd-catalyzed Suzuki-Miyaura reactions of dibromo sulfoxide and boronic acids, and the role of different phosphine ligands was also explored (Scheme 5) [112]. The calculations clarified that the regioselectivity is ascribed to different facility for ligand dissociation. For less bulky PPh3 ligand, it involves bisligated catalyst and prefers SN2 mechanism and α-bromosulfoxide activation. For bulky P(1-napthyl)3 ligand, it is related to the monoligated catalyst and proceeds via concerted mechanism and bromoaryl activation.

|
Download:
|
| Scheme 5. The C(sp2)-Br and C(sp3)-Br bonds activation by Pd catalysts with different phosphine ligands. | |
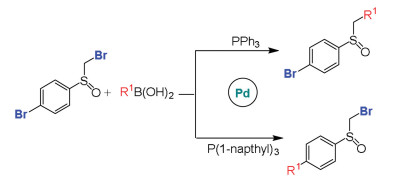{kind=link}
Fu et al. investigated the ligand-controlled regioselectivity in palladium-catalyzed Suzuki reactions of aryl boronic acids with chloro aryl triflate (Scheme 6) [113]. The tert-butylphosphine (PtBu3) ligand gave coupling at the C-Cl (C(sp2)-halides) bond, while the tricyclohexylphosphine (PCy3) ligand led to obvious reversal with the reaction at the C-OTf (C(sp2)-triflates) bond. Theoretical calculations on mechanistic details have been carried out by Houk and Schoenebeck to elucidate the origins for selectivity [114]. The calculations demonstrated that monocoordinate Pd(PtBu3) catalyst facilitates the C-Cl bond reaction, while the Pd(PCy3)2 catalyst prefers the C-OTf bond reaction [115, 116]. Distortion/interaction analysis elucidated that the O-C bond of triflate is difficult to distort compared to C-Cl bond. The C-Cl bond stretching can decrease the LUMO of substrate, which increases the interaction with palladium and further stabilizes this transition state, thus the regioselectivity is distortion-controlled. The orbital energies of TS indicated that bisligated palladium complex with high-lying HOMO is more nucleophilic, which can react at the C-OTf bond. The transition state with distorted geometry has a low-lying LUMO, therefore, this reaction displays the great interaction, and the regioselectivity is interaction-controlled.

|
Download:
|
| Scheme 6. The Suzuki reactions of chloro aryl triflate with aryl boronic acids by Pd catalysts with different phosphine ligands. | |
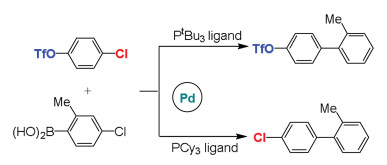{kind=link}
3.3.2. Activation of C–H bonds
The functionalization of C–H bond has attracted wide attention in organic synthesis, and great progresses have been made over the past decades [117-119]. Main challenges in palladium-catalyzed ligand-directed C–H activation is involved in the selectivity, and leads to many mechanistic investigations on different C–H bonds functionalization within a complex molecule [120-129]. Many different methods have been selected to solve this issue, and the common way is the use of substrates that contain coordinating ligands. Two different methods have been employed to ligand design. One is the coordinating ligands also called directing groups that bind to the Pd center, and selectively deliver the Pd center to activate a proximal C–H bond. The other is related to the isolated ligands. The common isolated ligands are mono-N-protected amino acid (MPAA) and acetylprotected aminoethyl quinoline (APAQ) ligands, which acts as the internal base. Computational studies are essential to the development of selective C–H bond activation, and understanding the origin for selectivity is necessary.
Davies and Macgregor reported the first theoretical calculations in Pd-catalyzed cyclometalation of dimethylbenzylamine by intramolecular C–H functionalization featuring a directing group [130], and this system has been widely investigated experimentally by Ryabov and co-workers (Fig. 4) [131]. The amine group of dimethylbenzylamine is the directing groups to make the catalysts selectively activate C–H bonds. From complex 1 [Pd(OAc)2(Me2NCH2Ph)], the reaction could proceed via two C–H activation processes. The one considers the displacement from κ2-OAc ligand to κ1-ortho-C–H bond of benzyl group through TS1, which gives an agostic complex 2 featuring a polarized C–H bond. The C–H bond cleavage occurs with a small barrier by TS2 to afford product 3.

|
Download:
|
| Fig. 4. The barriers (kcal/mol) for Pd-catalyzed cyclometalation of dimethylbenzylamine by intramolecular C–H functionalization. | |
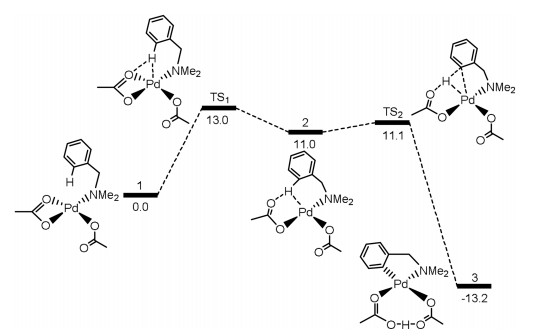{kind=link}
Fagnou et al. studied the palladium-catalyzed synthesis of dihydrobenzofurans by C(sp3)-H bond cleavage (1 → 2, Fig. 5) [132]. The reaction mechanism is related to concerted metalation deprotonation (CMD). From the model system [Pd(Ar)(OAc)(PMe3)], three possible C(sp3)-H bonds activation of 2-methylbutoxy substituent are proposed through concerted and inner-sphere deprotonation mechanism. This step proceeds by TS1 producing a six-membered palladacycle, and the barrier of this path is much lower than the C–H activation of ethyl CH2 (TS2) group or ethyl CH3 (TS3) group. The CMD mechanism is responsible for observed selectivity in experiments [133], and includes the reaction occurred at the 2 position of thiophene and ortho-C–H bond activation of pyridine N-oxide (Fig. 6) [134].

|
Download:
|
| Fig. 5. Pd-catalyzed synthesis of dihydrobenzofurans through C(sp3)-H bond cleavage (1 → 2) via three possible pathways. | |
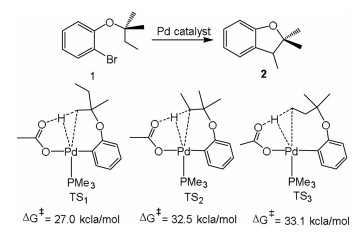{kind=link}

|
Download:
|
| Fig. 6. The barriers (kcal/mol) in Pd-catalyzed C–H bonds cleavage of heterocycles. | |
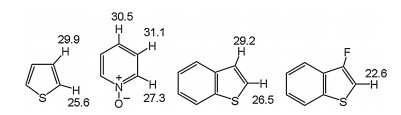{kind=link}
Clot and Baudoin reported the palladium-catalyzed β-arylation reaction of ester enolate with aryl bromide by experiments and computations [135, 136], and the ligand-controlled selectivity for α/β-arylation was explored (Scheme 7). For DavePhos ligand, the path via β-arylation is kinetically more favorable than that of α-arylation, and the isomerization from palladium–enolate to palladium-homoenolate is the rate-limiting step for β-arylation path. For PtBu3 ligand, the rate-limiting step is varied owing to the increasing barrier for isomerization, then the α-arylation of ester enolate is kinetically more favorable. In contrast to the other trialkylphosphine ligands, the relatively high activity of biaryldialkylphosphine ligands is ascribed to the stabilizing interactions between metal center and the biaryl backbone of ligand (TS in Scheme 7), thereby facilitates the β-arylation pathway.

|
Download:
|
| Scheme 7. Possible pathways for α/β-arylation. | |
{kind=link}
Houk and Yu discovered chiral acetyl-protected aminoethyl quinoline (APAQ) ligand that makes asymmetric Pd insert into prochiral C–H bond of methylene group (Fig. 7) [137]. The transition state TS_R is kinetically more favorable than TS_S, which is agree with the selectivity from experiments (90:10 er). The terminal methyl group of substrate for both transition states are oriented differently, and TS_S shows obvious substrate distortion, where the –CH3 group of substrate locates at the same face as α-aryl substituent of ligand and the peri-H of quinoline. This difference leads to the enantioselectivity.

|
Download:
|
| Fig. 7. Relative free energy barriers of two transition states for enantiomeric C–H bonds activation. | |
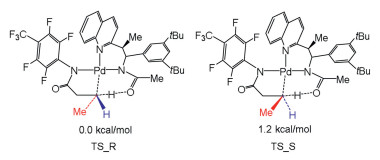{kind=link}
Fu et al. reported the theoretical calculations to demonstrate ligand-determined α- and β-arylation of thiophenes based on two possible mechanistic paths (Scheme 8) [138]. The 2, 2′-bipyridyl (bpy) ligand favors metalation/deprotonation path and leads to the α-arylated product, while the phosphite ligand facilitates Heck-type arylation to give the β-arylated product. The hydrogen bond interaction between the oxygen atom of carbonate and the hydrogen atom of bpy ligand is responsible for the β-arylation.

|
Download:
|
| Scheme 8. Different selectivity for the activation of C–H bonds of thiophene by Pd catalysts with different ligands. | |
{kind=link}
Houk and Yu carried out the DFT calculations to elucidate meta-selectivity in palladium- and silver-catalyzed activation of C–H bonds (Scheme 9a) [139]. The selectivity could be achieved via a Ag-Pd heterodimeric complex, where the nitrile group is coordinated to Ag center directs the meta-C–H to an acetate anion coordinated to Pd center. The role of bimetallic Ag-Pd complex was explored for ortho-C–H bond activation by Sunoj [140]. Zhang et al. also confirmed the facile of meta-C–H activation by DFT calculations (Scheme 9b) [141]. The calculations showed that steric repulsions between substrate and pyridine ligand favor meta-C–H bond activation, while electronic effects facilitate para-C–H bond activation.

|
Download:
|
| Scheme 9. Different C–H bonds activation by palladium catalyst. (a) The role of nitrile. (b) Steric repulsion between R group and ligand. | |
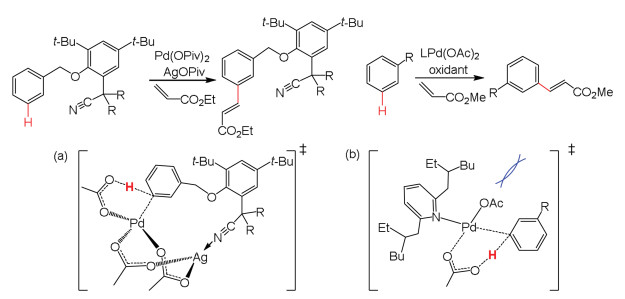{kind=link}
Wu et al. carried out IM-MS/DFT investigations on palladium-catalyzed enantioselective C–H bond activation with MPAA ligand [142]. The experiments indicated that [Pd(MPAA)] can activate substrate C–H bond without external bases, and MPAA ligand acting as internal base can accept a proton. Theoretical calculations were used to elucidate the conformational effect on enantioselectivity (Fig. 8). The MPAA ligand is coordinated to the palladium atom by bidentate mode, and the N-protecting group is seemed as internal base for selectivity-determining step. Selectivity is ascribed to the rigidity of MPAA ligand and substrate coordination, and steric repulsions is existed between the amino acid side-chain of N-protecting moiety and substrate, which make the H abstraction favorable via reasonable geometry [143].

|
Download:
|
| Fig. 8. The chirality relay from MPAA ligand to substrate. | |
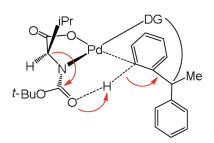{kind=link}
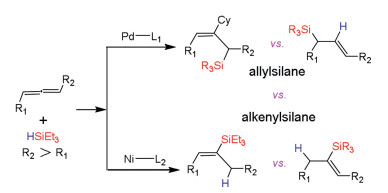
In general, nickel and palladium catalysts have the same catalytic role in cross-coupling reactions. Over the past years, many attentions have been paid to the differences between Pd and Ni catalysts. Our group reported the DFT computations on reaction mechanisms in palladium- and nickel-catalyzed hydrosilylation of mono-substituted allene, and the origins for regioselectivity have also been investigated (Scheme 10) [144]. The computations demonstrated that the Ni or Pd catalyst with different ligands may result from reverse selectivity for allene hydrosilylation. The nickel catalysts have lower barrier than that of palladium catalysts. Pd catalyst carrying small NHC ligand produces allylsilane, while Ni catalyst carrying bulky NHC ligand gives vinylsilane. The calculations elucidated that electronic and steric effects are significant to control reaction regioselectivity for allene hydrosilylation by palladium and nickel catalysts [145]. The substrate has been extended to disubstituted allene hydrosilylation by Pd and Ni catalysts [146, 147]. The DFT computations have performed to elucidate the reaction mechanism in Pd- and Ni-catalyzed 1, 3-disubstituted allene hydrosilylation, and the origins for regio- and stereo-selectivities have also been investigated (Scheme 11). The calculations suggested that the catalytic cycle contains three main steps, substrate coordination, concerted step involving oxidative addition commitant with hydride/silyl insertion, and C-Si/C–H reductive elimination. The results indicated that concerted step is the rate-determining and selectivity-controlling step for allene hydrosilylation. Furthermore, using palladium and nickel catalysts carrying NHC ligands may lead to switchable regioselectivity. The allene hydrosilylation reaction by nickel catalyst generates alkenylsilane, while the hydrosilylation of allene through Pd catalyst gives allylsilane. The electronic effects of palladium and nickel catalysts, and steric repulsions between substrates and catalysts control the reaction regioselectivity [146, 147].

|
Download:
|
| Scheme 10. The regioselectivity for mono-substituted allene hydrosilylation reaction by palladium and nickel catalysts. | |
{kind=link}

|
Download:
|
| Scheme 11. The regioselectivity for disubstituted allene hydrosilylation reactions. | |
{kind=link}
4. Summary and future perspectives
The wide applications of Pd- and Ni-catalyzed cross-coupling reactions are hot topic in academia and industry, and many calculations were performed to study the reaction mechanisms, while several challenges of explaining reaction selectivity remains.
Accurate modeling of free energies and reasonable chemical models are necessary to reflect complex experimental conditions, and molecular dynamics simulation has been used to organometallic systems, while this method requires a lot of computing resources and wide applications is thus limited. Furthremore, the conformational freedom of reactive species for transition metal systems is additional challenges [148], [149-150], and calculations require to solve different conformers arising from the flexible catalysts and ligands. The palladium and nickel catalysts carrying large ligands may lead to several possible conformations of reaction species and need to calculate individually. Force-field based method is seemed as a promising approach to deal with conformational space. Norrby et al. used Q2MM method to investigate the Trost ligands in Pd-catalyzed asymmetric reactions [151-153], and the parameters for transition-state-specific force field (TSFF) were fitted by quantum mechanistic data. The results proved that the Q2MM method is reasonable, quick and accurate to predict reaction selectivity. Previous studies indicated that the high-throughput screening of ligands is an effective and quick way to obtain expected selectivity [154-156], and this method depends on available ligand libraries. Recently, the parameterization of ligands has attracted extensive attentions [157, 158]. After preliminary screen of different ligands of nickel and palladium catalysts, it can quickly calculate descriptors on truncated ligands, and then builds the structure-selectivity relationship model to extrapolate into unknown ligand space [159].
This review introduced successful examples of computational explanation for the experimentally observed selectivity in Ni- and Pd-catalyzed coupling reactions, and we hope that calculations will be widely used before experiments to avoid wasteful experimental screening by quick computational evaluation.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21203166) and Visiting Scholars Project of Zhejiang Education Department (No. FX2017023).
| [1] |
T. Sperger, I.A. Sanhueza, I. Kalvet, F. Schoenebeck, Chem. Rev. 115 (2015) 9532-9586. DOI:10.1021/acs.chemrev.5b00163 |
| [2] |
S. Sakaki, Y.Y. Ohnishi, H. Sato, Chem. Rec. 10 (2010) 29-45. DOI:10.1002/tcr.200900019 |
| [3] |
M. García-Melchor, X. Solans-Monfort, G. Ujaque, C-C bond formation, in: J.R. Poeppelmeier (Ed. ), Comprehensive Inorganic Chemistry Ⅱ, Vol. 9, Elsevier, Amsterdam, 2013, pp. 767-805.
|
| [4] |
T. Fan, Z. Lin, Theoretical insights into transition metal-catalyzed reactions of carbon dioxide. Understanding organometallic reaction mechanisms and catalysis. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2014: pp. 121-144.
|
| [5] |
M. García-Melchor, A.A.C. Braga, A. Lledo's, G. Ujaque, F. Maseras, Acc. Chem. Res. 46 (2013) 2626-2634. DOI:10.1021/ar400080r |
| [6] |
K.J. Bonney, F. Schoenebeck, Chem. Soc. Rev. 43 (2014) 6609-6638. DOI:10.1039/C4CS00061G |
| [7] |
D. Balcells, E. Clot, O. Eisenstein, Chem. Rev. 110 (2010) 749-823. DOI:10.1021/cr900315k |
| [8] |
W.M.C. Sameera, F. Maseras, WIREs Comput. Mol. Sci. 2 (2012) 375-385. DOI:10.1002/wcms.1092 |
| [9] |
Y.Y. Jiang, Z. Li, J. Shi, Organometallics 31 (2012) 4356-4366. DOI:10.1021/om300329z |
| [10] |
D.G. Musaev, T.M. Figg, A.L. Kaledin, Chem. Soc. Rev. 43 (2014) 5009-5031. DOI:10.1039/C3CS60447K |
| [11] |
D. Balcells, O. Eisenstein, Theoretical studies on the reaction mechanism of metal-assisted C-H activation, in: J.R. Poeppelmeier (Ed. ), Comprehensive Inorganic Chemistry Ⅱ, Vol. 9, Elsevier, Amsterdam, 2013, pp. 695-726.
|
| [12] |
S.I. Gorelsky, Coord. Chem. Rev. 257 (2013) 153-164. DOI:10.1016/j.ccr.2012.06.016 |
| [13] |
L. Xue, Z. Lin, Chem. Soc. Rev. 39 (2010) 1692-1705. DOI:10.1039/B814973A |
| [14] |
H.J. Xie, T. Fan, Q.F. Lei, W.J. Fang, Sci. China Chem. 59 (2016) 1432-1447. DOI:10.1007/s11426-016-0018-2 |
| [15] |
F. Schoenebeck, Ligand, additive, and solvent effects in palladium catalysis-mechanistic studies en route to catalyst design. Understanding organometallic reaction mechanisms and catalysis. Germany: Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2014: pp. 69-92.
|
| [16] |
P. Liu, K.N. Houk, Inorg. Chim. Acta 369 (2011) 2-14. DOI:10.1016/j.ica.2010.12.042 |
| [17] |
H.J. Xie, Q. Sun, G.R. Ren, Z.X. Cao, J. Org. Chem. 79 (2014) 11911-11921. DOI:10.1021/jo501618k |
| [18] |
A.A.C. Braga, G. Ujaque, F. Maseras, Computational modeling for homogeneous and enzymatic catalysis, in: K. Morokuma, D.G. Musaev (Eds. ), A Knowledge-base for Designing Efficient Catalysis, WILEY-VCH GmbH & Co., KGaA, Weinheim, 2008, pp. 109-130.
|
| [19] |
G.B. Smith, G.C. Dezeny, D.L. Hughes, A.O. King, T.R. Verhoeven, J. Org. Chem. 59 (1994) 8151-8156. DOI:10.1021/jo00105a036 |
| [20] |
U. Christmann, R. Vilar, Angew. Chem. Int. Ed. 44 (2005) 366-374. DOI:10.1002/anie.200461189 |
| [21] |
M. Besora, C. Gourlaouen, B. Yates, F. Maseras, Dalton Trans 40 (2011) 11089-11094. DOI:10.1039/c1dt10983a |
| [22] |
H.M. Senn, T. Ziegler, Organometallics 23 (2004) 2980-2988. DOI:10.1021/om049963n |
| [23] |
E. Lyngvi, F. Schoenebeck, Tetrahedron 69 (2013) 5715-5718. DOI:10.1016/j.tet.2013.03.095 |
| [24] |
S. Kozuch, C. Amatore, A. Jutand, S. Shaik, Organometallics 24 (2005) 2319-2330. DOI:10.1021/om050160p |
| [25] |
L.J. Goossen, D. Koley, H.L. Hermann, W. Thiel, Organometallics 24 (2005) 2398-2410. DOI:10.1021/om0500220 |
| [26] |
J.N. Harvey, F. Himo, F. Maseras, L. Perrin, ACS Catal 9 (2019) 6803-6813. DOI:10.1021/acscatal.9b01537 |
| [27] |
N. Fey, M.F. Haddow, J.N. Harvey, C.L. McMullin, A.G.A. Orpen, Dalton Trans (2009) 8183-8196. |
| [28] |
J. Jover, N. Fey, J.N. Harvey, et al., Organometallics 29 (2010) 6245-6258. DOI:10.1021/om100648v |
| [29] |
J. Jover, N. Fey, J.N. Harvey, et al., Organometallics 31 (2012) 5302-5306. DOI:10.1021/om300312t |
| [30] |
Y.T. Zhang, M.N. Cao, G.Q. Ge, et al., Sci. Sin. Chim. 49 (2019) 380-390. |
| [31] |
H.J. Xie, F.R. Lin, Q.F. Lei, W.J. Fang, Organometallics 32 (2013) 6957-6968. DOI:10.1021/om400503x |
| [32] |
H.J. Xie, H. Zhang, Z.Y. Lin, Organometallics 32 (2013) 2336-2343. DOI:10.1021/om301215a |
| [33] |
H.J. Xie, H. Zhang, Z.Y. Lin, New J. Chem. 37 (2013) 2856-2861. DOI:10.1039/c3nj00531c |
| [34] |
H.J. Xie, F.R. Lin, L. Yang, et al., J. Organomet. Chem. 745-746 (2013) 417-422. DOI:10.1016/j.jorganchem.2013.08.015 |
| [35] |
H.J. Xie, F.R. Lin, Q.F. Lei, W.J. Fang, J. Phys. Org. Chem. 26 (2013) 933-938. DOI:10.1002/poc.3192 |
| [36] |
N. Fey, A.G. Orpen, J.N. Harvey, Coord. Chem. Rev. 253 (2009) 704-722. DOI:10.1016/j.ccr.2008.04.017 |
| [37] |
C.L. McMullin, J. Jover, J.N. Harvey, N. Fey, Dalton Trans 39 (2010) 10833-10836. DOI:10.1039/c0dt00778a |
| [38] |
N. Fey, B.M. Ridgway, J. Jover, C.L. McMullin, J.N. Harvey, Dalton Trans 40 (2011) 11184-11191. DOI:10.1039/c1dt10909j |
| [39] |
N. Fey, A.G. Orpen, J.N. Harvey, Coord. Chem. Rev. 253 (2009) 704-722. DOI:10.1016/j.ccr.2008.04.017 |
| [40] |
Y. Boutadla, D.L. Davies, S.A. Macgregor, A.I. Poblador-Bahamonde, Dalton Trans (2009) 5820-5831. |
| [41] |
C.J. Cramer, D.G. Truhlar, Acc. Chem. Res. 41 (2008) 760-768. DOI:10.1021/ar800019z |
| [42] |
R.F. Ribeiro, A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 115 (2011) 14556-14562. DOI:10.1021/jp205508z |
| [43] |
A.D. Becke, J. Chem. Phys. 98 (1993) 5648-5652. DOI:10.1063/1.464913 |
| [44] |
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785-789. DOI:10.1103/PhysRevB.37.785 |
| [45] |
T. Sperger, H.C. Fisher, F. Schoenebeck, WIREs Comput. Mol. Sci. 6 (2016) 226-242. DOI:10.1002/wcms.1244 |
| [46] |
T. Sperger, I.A. Sanhueza, F. Schoenebeck, Acc. Chem.Res. 49 (2016) 1311-1319. DOI:10.1021/acs.accounts.6b00068 |
| [47] |
M. Steinmetz, S. Grimme, ChemistryOpen 2 (2013) 115-124. DOI:10.1002/open.201300012 |
| [48] |
H.L. Woodcock, H.F. Schaefer Ⅲ, P.R. Schreiner, J. Phys. Chem. A 106 (2002) 11923-11931. DOI:10.1021/jp0212895 |
| [49] |
E.I. Izgorodina, M.L. Coote, L. Radom, J. Phys. Chem. A 109 (2005) 7558-7566. DOI:10.1021/jp052021r |
| [50] |
M.D. Wodrich, C. Corminboeuf, P.V.R. Schleyer, Org. Lett. 8 (2006) 3631-3634. DOI:10.1021/ol061016i |
| [51] |
P.R. Schreiner, A.A. Fokin, R.A. Pascal, A. de Meijere, Org. Lett. 8 (2006) 3635-3638. DOI:10.1021/ol0610486 |
| [52] |
M.D. Wodrich, C. Corminboeuf, P.R. Schreiner, A.A. Fokin, P.V.R. Schleyer, Org. Lett. 9 (2007) 1851-1854. DOI:10.1021/ol070354w |
| [53] |
S.E. Wheeler, A. Moran, S.N. Pieniazek, K.N. Houk, J. Phys. Chem. A 113 (2009) 10376-10384. DOI:10.1021/jp9058565 |
| [54] |
S. Grimme, J. Comput. Chem. 27 (2006) 1787-1799. DOI:10.1002/jcc.20495 |
| [55] |
T. Schwabe, S. Grimme, Acc. Chem. Res. 41 (2008) 569-579. DOI:10.1021/ar700208h |
| [56] |
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 132 (2010) 154104. DOI:10.1063/1.3382344 |
| [57] |
S. Grimme, WIREs Comput. Mol. Sci. 1 (2011) 211-228. DOI:10.1002/wcms.30 |
| [58] |
E. Lyngvi, I.A. Sanhueza, F. Schoenebeck, Organometallics 34 (2015) 805-812. DOI:10.1021/om501199t |
| [59] |
Y. Zhao, N.E. Schultz, D.G. Truhlar, J. Chem. Phys. 123 (2005) 161103. DOI:10.1063/1.2126975 |
| [60] |
Y. Zhao, N.E. Schultz, D.G. Truhlar, J. Chem. TheoryComput. 2 (2006) 364-382. |
| [61] |
Y. Zhao, D.G. Truhlar, Theor. Chem. Acc. 120 (2008) 215-241. DOI:10.1007/s00214-007-0310-x |
| [62] |
E. Lyngvi, I.A. Sanhueza, A. Schoenebeck, Organometallics 34 (2015) 805-812. DOI:10.1021/om501199t |
| [63] |
R. Martin, S.L. Buchwald, Acc. Chem. Res. 41 (2008) 1461-1473. DOI:10.1021/ar800036s |
| [64] |
C.A. Tolman, Chem. Rev. 77 (1977) 313-348. DOI:10.1021/cr60307a002 |
| [65] |
C.A. Tolman, W.C. Seidel, L.W. Gosser, J. Am. Chem. Soc. 96 (1974) 53-60. DOI:10.1021/ja00808a009 |
| [66] |
C.A. Tolman, J. Am. Chem. Soc. 92 (1970) 2953-2956. DOI:10.1021/ja00713a006 |
| [67] |
A.C. Hillier, W.J. Sommer, B.S. Yong, et al., Organometallics 22 (2003) 4322-4326. DOI:10.1021/om034016k |
| [68] |
A. Poater, B. Cosenza, A. Correa, et al., Eur. J. Inorg. Chem. (2009) 1759-1766. |
| [69] |
H. Clavier, S.P. Nolan, Chem. Commun. 46 (2010) 841-861. DOI:10.1039/b922984a |
| [70] |
X. Hong, P. Liu, K.N. Houk, J. Am. Chem. Soc. 135 (2012) 1456-1462. |
| [71] |
X. Wang, F. Meng, Y. Wang, et al., Angew. Chem. Int. Ed. 51 (2012) 9276-9282. DOI:10.1002/anie.201204925 |
| [72] |
E.W. Werner, T.S. Mei, A.J. Burckle, M.S. Sigman, Science 338 (2012) 1455-1458. DOI:10.1126/science.1229208 |
| [73] |
A.F. Littke, C. Dai, G.C. Fu, J. Am. Chem. Soc. 122 (2000) 4020-4028. DOI:10.1021/ja0002058 |
| [74] |
J. Mahatthananchai, J.W. Bode, Acc. Chem. Res. 47 (2014) 696-707. DOI:10.1021/ar400239v |
| [75] |
A. Gomez-Suarez, D.J. Nelson, S.P. Nolan, Chem. Commun. 53 (2017) 2650-2660. DOI:10.1039/C7CC00255F |
| [76] |
Z.D. Miller, W. Li, T.R. Belderrain, J. Montgomery, J. Am. Chem. Soc. 135 (2013) 15282-15285. DOI:10.1021/ja407749w |
| [77] |
E.I. Negishi, A. de Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis. New York: Wiley, 2002.
|
| [78] |
J.F. Hartwig, Organotransition Metal Chemistry-from Bonding to Catalysis, University Science Books. CA: Sausalito, 2010.
|
| [79] |
C.C.J. Seechurn, M.O. Kitching, T.J. Colacot, V. Snieckus, Angew. Chem. Int. Ed. 51 (2012) 5062-5085. DOI:10.1002/anie.201107017 |
| [80] |
M.C. Nielsen, K.J. Bonney, F. Schoenebeck, Angew. Chem. Int. Ed. 53 (2014) 5903-5906. DOI:10.1002/anie.201400837 |
| [81] |
R.B. Bedford, N.J. Gower, M.F. Haddow, et al., Angew. Chem. Int. Ed. 51 (2012) 5435-5438. DOI:10.1002/anie.201202219 |
| [82] |
C. Sköld, J. Kleimark, A. Trejos, et al., Chem. Eur. J. 18 (2012) 4714-4722. DOI:10.1002/chem.201102678 |
| [83] |
J. Jover, N. Fey, M. Purdie, G.C. Lloyd-Jones, J.N. Harvey, J. Mol. Catal. A 324 (2010) 39-47. DOI:10.1016/j.molcata.2010.02.021 |
| [84] |
Y. Ogiwara, Y. Sakurai, H. Hattori, N. Sakai, Org. Lett. 21 (2018) 4204-4208. |
| [85] |
T.Y. Xu, F. Sha, H. Alper, J. Am. Chem. Soc. 138 (2016) 6629-6635. DOI:10.1021/jacs.6b03161 |
| [86] |
Y. Zhu, S.L. Buchwald, J. Am. Chem. Soc. 136 (2014) 4500-4503. DOI:10.1021/ja501560x |
| [87] |
Q.Y. Meng, S. Wang, G.S. Huff, B. König, J. Am. Chem. Soc. 140 (2018) 3198-3201. DOI:10.1021/jacs.7b13448 |
| [88] |
X. Lin, J. Sun, Y. Xi, D. Lin, Organometallics 30 (2011) 3284-3292. DOI:10.1021/om1012049 |
| [89] |
Q. Lu, H. Yu, Y. Fu, J. Am. Chem. Soc. 136 (2014) 8252-8260. DOI:10.1021/ja4127455 |
| [90] |
K. Amaike, K. Muto, J. Yamaguchi, K. Itami, J. Am. Chem. Soc. 134 (2012) 13573-13576. DOI:10.1021/ja306062c |
| [91] |
K. Muto, J. Yamaguchi, K. Itami, J. Am. Chem. Soc. 134 (2012) 169-172. DOI:10.1021/ja210249h |
| [92] |
S.O. Nilsson Lill, P. Ryberg, T. Rein, E. Bennström, P.O. Norrby, Chem. Eur. J. 18 (2012) 1640-1649. DOI:10.1002/chem.201102674 |
| [93] |
X. Hong, Y. Liang, K.N. Houk, J. Am. Chem. Soc. 136 (2014) 2017-2025. DOI:10.1021/ja4118413 |
| [94] |
H.J. Xie, Y. Li, L.H. Wang, et al., Dalton Trans. 46 (2017) 13010-13019. DOI:10.1039/C7DT02532G |
| [95] |
S.Q. Zhang, B.L.H. Taylor, C.L. Ji, et al., J. Am. Chem. Soc. 139 (2017) 12994-13005. DOI:10.1021/jacs.7b04973 |
| [96] |
Z. Li, S.L. Zhang, Y. Fu, Q.X. Guo, L. Liu, J. Am. Chem. Soc. 131 (2009) 8815-8823. DOI:10.1021/ja810157e |
| [97] |
A. Chatupheeraphat, H.H. Liao, W. Srimontree, et al., J. Am. Chem. Soc. 140 (2018) 3724-3735. DOI:10.1021/jacs.7b12865 |
| [98] |
H.A. Malik, G.J. Sormunen, J.A. Montgomery, J. Am. Chem. Soc. 132 (2010) 6304-6305. DOI:10.1021/ja102262v |
| [99] |
P. Liu, K.N. Houk, Inorg. Chimi. Acta 369 (2011) 2-14. DOI:10.1016/j.ica.2010.12.042 |
| [100] |
P.R. McCarren, P. Liu, P.H.Y. Cheong, T.F. Jamison, K.N. Houk, J. Am. Chem. Soc. 131 (2009) 6654-6655. DOI:10.1021/ja900701g |
| [101] |
R.M. Moslin, K. Miller-Moslin, T.F. Jamison, Chem. Commun. (2007) 4441-4449. |
| [102] |
P. Liu, J. Montgomery, K.N. Houk, J. Am. Chem. Soc. 133 (2011) 6956-6959. DOI:10.1021/ja202007s |
| [103] |
H.B. Wang, G. Lu, G.J. Sormunen, et al., J. Am. Chem. Soc. 139 (2017) 9317-9324. DOI:10.1021/jacs.7b04583 |
| [104] |
V.H.M. da Silva, A.P.L. Batista, O. Navarro, A.A.C. Braga, J. Comput. Chem. 38 (2017) 2371-2377. DOI:10.1002/jcc.24867 |
| [105] |
S.B. Xu, J.L. Jiang, L.L. Ding, Y. Fu, Z.H. Gu, Org. Lett. 20 (2018) 325-328. DOI:10.1021/acs.orglett.7b03514 |
| [106] |
L.J. Liu, G.J. Pei, P. Liu, et al., J. Org. Chem. 83 (2018) 2067-2076. DOI:10.1021/acs.joc.7b03007 |
| [107] |
C.L. McMullin, N. Fey, J.N. Harvey, Dalton Trans 43 (2014) 13545-13556. DOI:10.1039/C4DT01758G |
| [108] |
F. Hartwig, Pure Appl. Chem. 71 (1999) 1417-1423. DOI:10.1351/pac199971081417 |
| [109] |
B. Pudasaini, B.G. Janesko, Organometallics 31 (2012) 4610-4618. DOI:10.1021/om300455g |
| [110] |
N. Rodríguez, C. Ramírez de Arellano, G. Asensio, M. Medio-Simo'n, Chem. Eur. J. 13 (2007) 4223-4229. DOI:10.1002/chem.200601488 |
| [111] |
C. Gourlaouen, G. Ujaque, A. Lledo's, et al., J. Org. Chem. 74 (2009) 4049-4054. DOI:10.1021/jo900178c |
| [112] |
C. Mollar, M. Besora, F. Maseras, G. Asensio, M. Medio-Simo'n, Chem. Eur. J. 16 (2010) 13390-13397. DOI:10.1002/chem.201001113 |
| [113] |
A.F. Littke, C. Dai, G.C. Fu, J. Am. Chem. Soc. 122 (2000) 4020-4028. DOI:10.1021/ja0002058 |
| [114] |
F. Proutiere, F. Schoenebeck, Angew. Chem. Int. Ed. 50 (2011) 8192-8195. DOI:10.1002/anie.201101746 |
| [115] |
F. Proutiere, E. Lyngvi, M. Aufiero, I.A. Sanhueza, F. Schoenebeck, Organometallics 33 (2014) 6879-6884. DOI:10.1021/om5009605 |
| [116] |
F. Schoenebeck, K.N. Houk, J. Am. Chem. Soc. 132 (2010) 2496-2497. DOI:10.1021/ja9077528 |
| [117] |
R. Giri, B.F. Shi, K.M. Engle, N. Maugel, J.Q. Yu, Chem. Soc. Rev. 38 (2009) 3242-3272. DOI:10.1039/b816707a |
| [118] |
M.E. O'Reilly, R. Fu, R.J. Nielsen, et al., J. Am. Chem. Soc. 136 (2014) 14118-14126. |
| [119] |
T. Higashi, H. Ando, S. Kusumoto, K. Nozaki, J. Am. Chem. Soc. 141 (2019) 2247-2250. DOI:10.1021/jacs.8b13829 |
| [120] |
T.W. Lyons, M.S. Sanford, Chem. Rev. 110 (2010) 1147-1169. DOI:10.1021/cr900184e |
| [121] |
L. Theveau, O. Querolle, G. Dupas, C. Hoarau, Tetrahedron 69 (2013) 4375-4380. DOI:10.1016/j.tet.2013.01.073 |
| [122] |
Q. Zhang, H.Z. Yu, Y. Fu, Organometallics 32 (2013) 4165-4173. DOI:10.1021/om400370v |
| [123] |
R. Meir, S. Kozuch, A. Uhe, S. Shaik, Chem. Eur. J. 17 (2011) 7623-7631. DOI:10.1002/chem.201002724 |
| [124] |
D.E. Stephens, J. Lakey-Beitia, A.C. Atesin, et al., ACS Catal. 5 (2015) 167-175. DOI:10.1021/cs501813v |
| [125] |
S. Rousseaux, M. Davi, J. Sofack-Kreutzer, et al., J. Am. Chem. Soc. 132 (2010) 10706-10716. DOI:10.1021/ja1048847 |
| [126] |
C.E. Kefalidis, M. Davi, P.M. Holstein, E. Clot, O. Baudoin, J. Org. Chem. 79 (2014) 11903-11910. DOI:10.1021/jo501610x |
| [127] |
S.Y. Tang, Q.X. Guo, Y. Fu, Chem. Eur. J. 17 (2011) 13866-13876. DOI:10.1002/chem.201101587 |
| [128] |
R. Giri, Y. Lan, P. Liu, K.N. Houk, J.Q. Yu, J. Am. Chem. Soc. 134 (2012) 14118-14126. DOI:10.1021/ja304643e |
| [129] |
M. Steinmetz, K. Ueda, S. Grimme, et al., Chem. Asian J. 7 (2012) 1256-1260. DOI:10.1002/asia.201101011 |
| [130] |
D.L. Davies, S.M.A. Donald, S.A. Macgregor, J. Am. Chem. Soc. 127 (2005) 13754-13755. DOI:10.1021/ja052047w |
| [131] |
A.D. Ryabov, I.K. Sakodinskaya, A.K. Yatsimirsky, Chem J., Soc., Dalton Trans. (1985) 2629-2638. |
| [132] |
M. Lafrance, S.I. Gorelsky, K. Fagnou, J. Am. Chem. Soc. 129 (2007) 14570-14571. DOI:10.1021/ja076588s |
| [133] |
S.I. Gorelsky, D. Lapointe, K. Fagnou, J. Am. Chem. Soc. 130 (2008) 10848-10849. DOI:10.1021/ja802533u |
| [134] |
D. Lapointe, T. Markiewicz, C.J. Whipp, A. Toderian, K. Fagnou, J. Org. Chem. 76 (2011) 749-759. DOI:10.1021/jo102081a |
| [135] |
P. Larini, C.E. Kefalidis, R. Jazzar, et al., Chem. Eur. J. 18 (2012) 1932-1944. DOI:10.1002/chem.201103153 |
| [136] |
A. Millet, P. Larini, E. Clot, O. Baudoin, Chem. Sci. 4 (2013) 2241-2247. DOI:10.1039/c3sc50428j |
| [137] |
G. Chen, W. Gong, Z. Zhuang, et al., Science 353 (2016) 1023-1027. DOI:10.1126/science.aaf4434 |
| [138] |
S.Y. Tang, Q.X. Guo, Y. Fu, Chem. Eur. J. 17 (2011) 13866-13876. DOI:10.1002/chem.201101587 |
| [139] |
Y.F. Yang, G.J. Cheng, P. Liu, et al., J. Am. Chem. Soc. 136 (2014) 344-355. DOI:10.1021/ja410485g |
| [140] |
M. Anand, R.B. Sunoj, H.F. Schaefer, J. Am. Chem. Soc. 136 (2014) 5535-5538. DOI:10.1021/ja412770h |
| [141] |
S. Zhang, L. Shi, Y. Ding, J. Am. Chem. Soc. 133 (2011) 20218-20229. DOI:10.1021/ja205294y |
| [142] |
G.J. Cheng, P. Chen, T.Y. Sun, et al., Chem. Eur. J. 21 (2015) 11180-11188. DOI:10.1002/chem.201501123 |
| [143] |
Y.F. Yang, X. Hong, J.Q. Yu, K.N. Houk, Acc. Chem. Res. 50 (2017) 2853-2860. DOI:10.1021/acs.accounts.7b00440 |
| [144] |
H.J. Xie, L.J. Zhao, Y. Yang, et al., J. Org. Chem. 79 (2014) 4517-4527. DOI:10.1021/jo500557w |
| [145] |
Z.D. Miller, W. Li, T.R. Belderrain, J. Montgomery, J. Am. Chem. Soc. 135 (2013) 15282-15285. DOI:10.1021/ja407749w |
| [146] |
Z.D. Miller, R. Dorel, J. Montgomery, Angew. Chem. Int. Ed. 54 (2015) 9088-9091. DOI:10.1002/anie.201503521 |
| [147] |
H.J. Xie, J. Kuang, L.H. Wang, et al., Organometallics 36 (2017) 3371-3381. DOI:10.1021/acs.organomet.7b00495 |
| [148] |
M. Burns, S. Essafi, J.R. Bame, et al., Nature 513 (2014) 183-188. DOI:10.1038/nature13711 |
| [149] |
S. Maeda, K. Ohno, K. Morokuma, Phys. Chem. Chem. Phys. 15 (2013) 3683-3701. DOI:10.1039/c3cp44063j |
| [150] |
A.L. Dewyer, A.J. Arguelles, P.M. Zimmerman, WIREs Comput. Mol. Sci. 8 (2018) No. e1354. DOI:10.1002/wcms.1354 |
| [151] |
C. Butts, E. Filali, G. Lloyd-Jones, et al., J. Am. Chem. Soc. 131 (2009) 9945-9957. DOI:10.1021/ja8099757 |
| [152] |
P.J. Donoghue, P. Helquist, P.O. Norrby, O. Wiest, J. Chem. Theory Comput. 4 (2008) 1313-1323. DOI:10.1021/ct800132a |
| [153] |
P.J. Donoghue, P. Helquist, P.O. Norrby, O. Wiest, J. Am. Chem. Soc. 131 (2009) 410-411. DOI:10.1021/ja806246h |
| [154] |
K.C. Harper, M.S. Sigman, Science 333 (2011) 1875-1878. DOI:10.1126/science.1206997 |
| [155] |
K.C. Harper, E.N. Bess, M.S. Sigman, Nat. Chem. 4 (2012) 366-374. DOI:10.1038/nchem.1297 |
| [156] |
K.C. Harper, M.S. Sigman, J. Org. Chem. 78 (2013) 2813-2818. DOI:10.1021/jo4002239 |
| [157] |
K. Wu, A.G. Doyle, Nat. Chem. 9 (2017) 779-784. DOI:10.1038/nchem.2741 |
| [158] |
Z.L. Niemeyer, A. Milo, D.P. Hickey, M.S. Sigman, Nat. Chem. 8 (2018) 610-617. |
| [159] |
M.H. Keylor, Z.L. Niemeyer, M.S. Sigman, K.L. Tan, J. Am. Chem. Soc. 139 (2017) 10613-10616. DOI:10.1021/jacs.7b05409 |