 2021, Vol. 32
2021, Vol. 32 


b Department of Applied Chemistry, Graduate School of Engineering, Hiroshima University, Higashi-Hiroshim, 739-8527, Japan;
c Beijing Key Laboratory of Flavour Chemistry, Beijing Technology and Business University (BTBU), Beijing 100048, China
 Jiajing Tan obtained his BS from University of Science and Technology of China in 2008. He received his PhD in 2013 under the supervision of Professor Hisashi Yamamoto at the University of Chicago. From 2013–2015, he worked at Merck & Co. Then, he started his academic career at Beijing University of Chemical and Technology. His current research interests are aryne chemistry and green chemistry.
Jiajing Tan obtained his BS from University of Science and Technology of China in 2008. He received his PhD in 2013 under the supervision of Professor Hisashi Yamamoto at the University of Chicago. From 2013–2015, he worked at Merck & Co. Then, he started his academic career at Beijing University of Chemical and Technology. His current research interests are aryne chemistry and green chemistry. |
Organosulfur compounds are widely present in both natural products and active pharmaceutical ingredients [1]. According to statistical results, 24.8% of the top 200 brand name drugs by U.S. retail sales contain the sulfur atoms [2]. Besides, sulfur-based compounds are versatile synthetic reagents and play an important role in the drug discovery process [3]. The widespread application of sulfur-containing compounds could be attributed to its large size and lower electronegativity, which enable the outer electron shell of sulfur prone to either accept or donate electrons. In this case, the valence state of sulfur can range from -2 to +6, and thus display unique reactivities when compared to oxygen [1].
Sulfonium salt is defined as positively charged sulfur carrying three carbon-sulfur bonds, while sulfur ylide possesses a formally negatively charged atom attaching to the sulfonium. Both of them are important S(Ⅳ) species. Due to significant application profiles and unique chemical reactivities of organosulfur compounds, developing more efficient synthesis methods while discovering new reactivities has still been a challenging yet elusive task in this field [4]. Such synthetic campaigns could be dated back to 1960, when Johnson, Corey and Chaykovsky independently developed the reaction of sulfur ylides with carbonyls or imines to synthesize three-membered ring compounds [5]. From then on, numerous efforts have been made, many of which were documented as named reactions [6] including Pummerer reaction, Doyle-Kirmse reaction etc. In particular, sulfonium salts and sulfur ylides with unique reactivities have drawn great attentions as it could provide simple, effective, and often stereoselective methods to gain access to versatile sulfur-containing structural motifs. Research performed during the past several years has been impressively fruitful in unveiling new aspects of sulfonium and sulfur ylide chemistry, which have become one of the major focuses for synthetic chemists [7]. In light of recent advances, it would be timely to provide a comprehensive review of the plethora of transformations within this field.
Sulfonium salt and sulfur ylide are one of the most important class of organosulfur(Ⅳ) species. In the following sections, we summarize and unify the current knowledge, with special emphasis on the significant development of sulfonium salt and sulfur ylide chemistry from Jan 2016 to Dec 2019. Ground-breaking reports related to this topic are included occasionally with the purpose of providing a background introduction. The critical goal of our review is to draw attention to this burgeoning research area, and stimulate further interests from the synthetic community in discovering new reactivities as well as multidisciplinary applications. Literature reports either from the same group or on the same topic but contributed by different groups were summarized together. For the sake of convenience, the review is organized into three sections based on the role of sulfonium salt and sulfur ylide play in organic reactions (synthetic reagents, intermediates and catalysts).
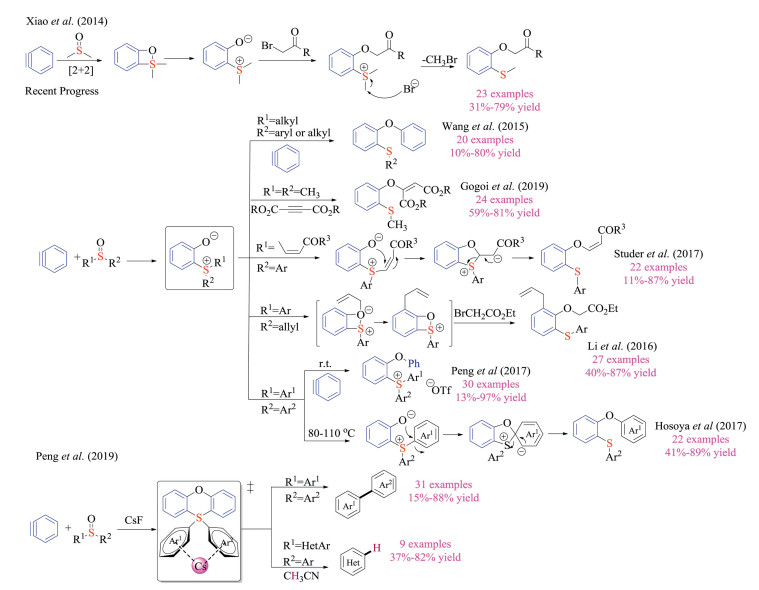
2. As reactantsAs important S(Ⅳ) species, certain sulfonium salts, and sulfur ylides have been employed as stable substrates in organic synthesis upon appropriate activations. Recent progress has been summarized as follows.
2.1. Sulfonium salts coupling partners in metal catalyzed reactionsTransition metal catalyzed coupling reactions are one of the most widely utilized reactions in the pharmaceutical industry due to the mild reaction conditions, functional group compatibility and scale-up robustness [8]. Sulfonium salts are well-recognized for their good stability, low toxicity and resulting operational simplicity, which in principle could be employed as reagents in organic synthesis. Besides, sulfonium salts displayed great solubility in both aprotic and protic solvents. Given these properties, the Lisbeskind group in 1997 reported the pioneering work of employing sulfonium salts as cationic coupling partner in palladium or Nickel catalyzed carbon-carbon bond formation reactions (Scheme 1) [9]. However, only until recently has this research topic attracted widespread attentions.

|
Download:
|
| Scheme 1. Lisbeskind's pioneering work. | |
{kind=link}
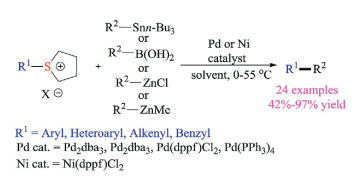
Inspired by this pioneering report, Zhang and co-workers have systematically researched on employing sulfonium salts as substrates in metal-catalyzed cross-coupling reactions (Scheme 2) [10]. In 2016, they reported a Pd-catalyzed Mizoroki-Heck-type reaction of Yagupolskii-Umemoto reagents at room temperature with alkenes [10a]. The coupling of both conjugated and unconjugated alkenes proceeded smoothly to give coupled products in moderate to good yields. Interestingly, toluenesulfonic acids as additives proved to be effective to promote the reaction, while traditional Heck reactions often needed bases to neutralize the in-situ formed acids and facilitated the ligand recovery. Later, they also successfully developed the Suzuki-Miyaura cross-coupling reactions of Yagupolskii-Umemoto reagents [10b]. Mechanistic study further confirmed that the C(sp2)-S bond could be effectively cleaved by Pd complexes. Investigations on substitution group effects suggested that the relative electron-poor aryl groups on sulfonium salts be prone to undergo arylation reactions than electron-rich ones. In 2017, the same group extended the application to Sonogashira couplings [10c]. Triaryl-, alkyl(diaryl)-, and aryl(dialkyl)-sulfonium salts were all discovered to be viable substrates, delivering target coupling products in high yields. In 2019, they finally achieved the C-N coupling reaction of alkyl(aryl)-sulfonium triflates. Under the palladium/copper co-catalysis, the Caryl-S bond readily cleaved to react with anilines [10d]. In fact, these protocols not only demonstrated the feasibilities that sulfonium salts could function as an alternative class of cross-coupling partners, but also provided important insights into the reactivity of sulfonium salts in transition metal catalysis.

|
Download:
|
| Scheme 2. Zhang's systematic work on cross-coupling of sulfonium salts. | |
{kind=link}
Yorimitsu group also focused on employing sulfonium salts as the synthetic equivalents of aryl halides in transition metal-catalyzed cross-coupling reaction. In addition to the carbon-carbon bond formations [11a], Yorimitsu et al. reported the first example of palladium-catalyzed direct borylation reaction of aryl sulfonium salts with B2pin2 under mild conditions, using palladium acetate as the catalyst and X-Phos as the ligand (Scheme 3) [11b]. Since the aryl sulfonium substrates were easily prepared through methylation of thioethers, aryl sulfide substrates could then be transformed into corresponding borylation products even in a one-pot manner. The methylation reactions occurred predominantly at the more nucleophilic sulfides, which in turn were challenging for the oxidative addition of metal catalysts. Thus, the overall coupling achieved selective borylation on sulfur motifs over chloro- and tosyloxy-groups, which should offer orthogonality for synthetic applications. Later, the alkoxycarbonylation reaction of aryl sulfonium salts was realized by the same group using the palladium-Xantphos system under the atmospheric pressure of CO gas [11c]. Various functional groups including carbonyl, cyano, halo, and sulfonyl groups were well-tolerated. The one-pot procedure was also developed to enable the direct transformation of aryl methyl sulfides to aryl esters.

|
Download:
|
| Scheme 3. Yorimitsu's research on cross-coupling of sulfonium salts. | |
{kind=link}
In 2018, Novák and co-workers employed sulfonium salts as suitable alkylation reagents in palladium-catalyzed C–H activation reactions (Scheme 4) [12]. The direct ortho-alkylation of anilides and aromatic ureas gave coupled products with good yields in the presence of palladium catalysts. The trifluoroacetic acid additive completely inhibited the N-alkylation by-products, while the addition of copper acetate significantly improved the reaction outcomes. Compared to electrophilic iodonium salts, these simply prepared sulfonium salts were considered to be new reagents in such type of transformations, and hopefully, their unique reactivities could open new paths in the field of transition-metal-catalyzed C–H activation.

|
Download:
|
| Scheme 4. Sulfonium salts as alkylation reagents in C-H activation reactions. | |
{kind=link}
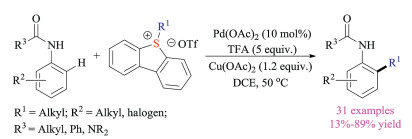
The above-mentioned reactions mainly relied on the nucleophilic substitution reaction of thioethers with alkylation reagents to prepare sulfonium salts. In fact, such alkyl halide reagents were often toxic and difficult to handle. Recently, Cowper and Lewis reported another breakthrough in this field. They creatively used Pummerer type reaction to synthesize azulene sulfonium salts and successfully introduced these bench-stable crystalline species as pseudo-halides for palladium-catalyzed Suzuki-Miyaura cross-coupling reactions (Scheme 5) [13]. Compared to corresponding halide species, the azulene sulfonium salts were not only easy to prepare, but also stable for purification as well as operation. Shortly after, Procter group also disclosed a sequential interrupted Pummerer and nickel-catalyzed cross-coupling strategy [14]. This protocol employed sulfoxides to activate styrene via direct formation of stable sulfonium salts as electrophilic coupling partners, which then participated in the nickel-catalyzed Negishi type reaction with arylzinc reagents. Indeed, this reaction could be performed in a one-pot fashion to give highly functionalized styrene products in good yields. Moreover, carbo- and heterocyclic alkenyl sulfonium salt substrates, which proved to be relatively difficult to synthesize via other routes, could be easily obtained via interrupted Pummerer reactions.

|
Download:
|
| Scheme 5. Tandem Pummerer and coupling reaction. | |
{kind=link}
In these above-mentioned metal-catalyzed cross-coupling reactions, the carbon-sulfur bond in sulfonium salts proved to be efficient for cleavage under transition metal catalysis, which is probably due to their inherent electron-deficient nature. Moreover, the neutral sulfide group left from sulfonium salts ought to be less poisonous for metal catalysts, when comparing to anionic sulfur species.
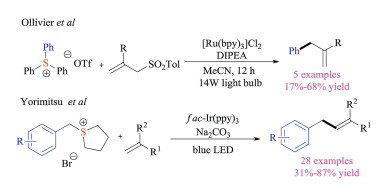
2.2. Photochemical reactionsDuring the past two decades, the renaissance of radical chemistry enabled resurgence in the interests of photochemistry, which has gradually emerged as an important weapon for synthetic chemists [15]. Sulfonium salts could also be employed as substrates in photoredox reactions due to their low reduction potentials [16]. Back to 2013, Ollivier and colleagues reported that triarylsulfonium salts, commonly used as photoacid generators in polymeric chemistry, could produce aryl radicals under visible light-based reduction conditions through single electron transfer and subsequent C–S bond fragmentation (Scheme 6) [17]. The transient aryl radicals could be trapped with either allyl sulfones or 1, 1-diphenylethylenes to give corresponding arylated products. Recently, Yorimitsu group also developed a fac-Ir(ppy)3 catalyzed photoredox alkenylation reaction of benzylsulfoniums, giving the desired allylbenzene products in good chemical yields [18]. A variety of functional groups that are typically not amenable in metal catalyzed coupling reactions, such as halogen, ester, and cyano groups, were well tolerated in this radical transformation.

|
Download:
|
| Scheme 6. Sulfonium salts as aryl radical precursors under photochemical conditions. | |
{kind=link}
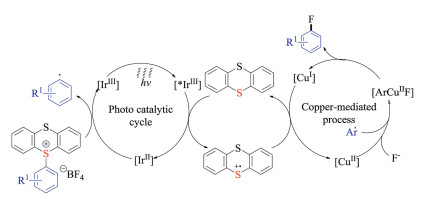
Ritter and co-workers recently succeeded in developing a highly site-selective C–H functionalization reaction of aromatic compounds (Scheme 7) [19a]. Instead of using directing groups or certain substitution patterns, this novel protocol relied on a persistent sulfur-based radical that could discriminate many C–H bonds even in a complex small molecule. More importantly, the resulting thianthrenium salts served as a powerful linchpin, which subjected to a variety of photochemical transformations as well as the above-mentioned metal-catalyzed cross-coupling reactions. In consistent with previous reports, thianthrenium salts were cleaved into thianthrenes and aryl radicals after single-electron reduction during photocatalysis. In fact, the high regioselectivities brought by the C–H thianthrenation strategy should render great opportunities for developing late stage C–H functionalization methodologies.

|
Download:
|
| Scheme 7. Ritter's pioneering work on C-H functionalization of aromatic compounds. | |
{kind=link}
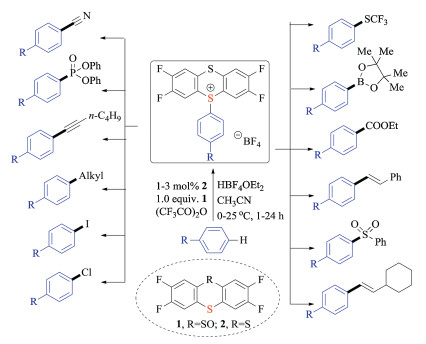
Based on this pioneering discovery, the same group also demonstrated the advantages of aryl thianthrenium salts for metallophotoredox catalysis [19b]. In the presence of an iridium(Ⅲ) photocatalyst and a copper(Ⅰ) redox catalyst, a variety of aryl fluorides were prepared in good efficiencies (Scheme 8). Again, the regioselective C–H thianthrenation strategy made the late-stage aromatic fluorination feasible. Substrates that were tolerated in either palladium-catalyzed C–F cross-couplings or electrophilic fluorination reactions, proved to be amenable under these reported conditions. Owing in part to the positive charge on sulfur, these sulfonium salts turned out to be relatively easier to reduce than corresponding aryl halides. As a result, many new photochemical transformations have been discovered accordingly.

|
Download:
|
| Scheme 8. Fluorination of sulfonium salts via metallophotoredox catalysis. | |
{kind=link}
The applications of thianthrenium salts as precursors were then extended to the C-O coupling, C-N coupling as well as the installation of trifluoromethyl groups (Scheme 9) [19c–e]. These protocols were not only scalable, but also displayed broad functional group tolerance, setting base for the functionalization of complex small molecules at late stage with high selectivities.

|
Download:
|
| Scheme 9. Ritter's follow-up work on thianthrenium salts. | |
{kind=link}
Recently, Gao, Du, Tang and co-workers developed an alternative borylation reaction of aryl sulfonium salts under photo-irradiation conditions (Scheme 10) [20]. A large array of aromatic boronate esters could be prepared in moderate to good yields. In this case, UV-irradiation (254 nm) was employed to excite simple aryl sulfonium salts to achieve C–S bond cleavage to form aryl radicals. Meanwhile, pyridine could bind to the boron center of B2Pin2 and provide the resulting activated borylation complex. It is worth mentioning that this approach was not applicable to benzyl and alkyl sulfonium salts, as the decomposition including demethylation and cyclization reaction took place rapidly. Although increasing number of research works on applications of sulfonium salts in photochemistry were reported, their utilization in organic electrochemistry was seldom explored.

|
Download:
|
| Scheme 10. Borylation of sulfonium salts under photochemical conditions. | |
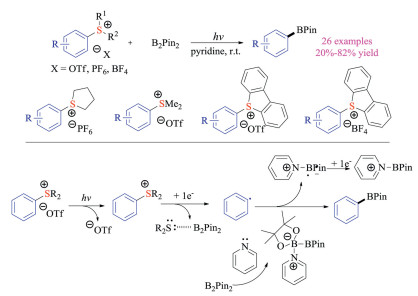{kind=link}
2.3. SNAr reactions
Nucleophilic aromatic substitution (SNAr) reactions are an old discipline with a rich history of synthetic applications [21]. Lately, many new advances have been on the horizon in order to match growing multidisciplinary research purposes. For instance, positron emission tomography (PET) techniques have recently emerged as one of the most effective molecular imaging tools to visualize biochemical processes in vivo, which often required labelling target with positron-emitting radioisotope such as 18F-fluorides [22]. Due to robustness and operation simplicity, SNAr reactions of sulfonium salts have become one of the most widely-used methods for incorporating 18F-fluorides [23].
In 2018, Årstad group reported a novel intramolecular ring-closing reaction to synthesize highly functionalized dibenzothiophene sulfonium salts from simple biaryl thioethers under mild conditions (Scheme 11) [24]. The cyclic sulfonium salt could further react with 18F-fluorides regioselectivity to give desired labeled products, and the obtained radiochemical yields were in correlation closely with theoretical DFT calculations. Moreover, the utilities of this protocol were further demonstrated by the radiolabeling of medicinally relevant compounds.

|
Download:
|
| Scheme 11. Aromatic 18F-fluorination of dibenzothiophene sulfonium salts. | |
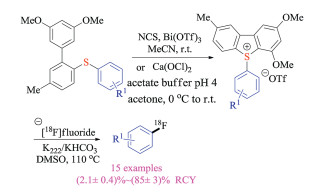{kind=link}
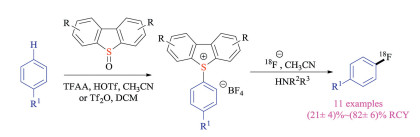
Recently, Ritter and co-workers also developed a site-selective late-stage aromatic 18F-fluorination protocol via SNAr pathway (Scheme 12) [25]. Similarly, the reported strategy relied on the site-selective aromatic C–H activation reaction to enable the formation of key sulfonium salt substrates. In this case, the arylthianthrenium salts worked best in previous reports [19] gave the undesired fluorinated aryl thioethers products. Instead, aryl dibenzothiophenium salts could undertake the desired SNAr reaction to give aryl fluorides using simple inorganic fluoride salts. Moreover, aryldibenzothiophenium salts could be utilized directly for elution of 18F-fluoride without adding of extra bases or kryptofix.

|
Download:
|
| Scheme 12. Fluorination of aryldibenzothiophenium salts. | |
{kind=link}
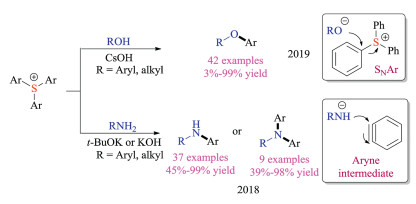
In addition to coupling reactions, Zhang group also developed the transition metal-free O-arylation reaction of triarylsulfonium triflates using alcohols and phenols as nucleophiles (Scheme 13) [26a]. This SNAr protocol displayed great substrate scope and corresponding C-O coupled products were obtained in good yields. Interestingly, the thianthrenium and dibenzothiophenium showed a similar reactivity trend as Ritter's fluorination work mentioned above [25]. They also discovered that triarylsulfonium salts were applicable for transition metal-free N-arylation reactions [26b]. However, deuterium labelling experiments suggested that aryne intermediates was generated from sulfonium salts in this case, which was also supported by the regioisomers obtained for 4-substituted triarylsulfonium salts. In the presence of a strong inorganic base (t-BuOK or KOH), a wide range of primary and secondary amines were readily converted to corresponding N-arylation products in good yields.

|
Download:
|
| Scheme 13. Zhang's work on nucleophilic aromatic substitution reaction. | |
{kind=link}
Later, Zhang, Wang and co-workers also demonstrated the nucleophilic aromatic substitution reaction of aryldimethyl sulfonium salts (Scheme 14) [27]. Various nucleophiles (including O, S, Se, Sn, Si centered nucleophiles) were well tolerated under standard conditions to give target products in moderate to good yields at room temperatures. Although transition metals were avoided, certain electron-withdrawing substituent groups on the aromatic rings were still required to offer better reactivities for C–S bond cleavage of arylsulfonium salts. Notably, a number of natural products and bioactive molecules could be utilized as nucleophiles for this transformation.

|
Download:
|
| Scheme 14. SNAr reaction of sulfonium salts. | |
{kind=link}
2.4. As fluoroalkylation reagents and other electrophilic reagents
When the anion in sulfur ylide was delocalized by one or more electron-withdrawing group, the stability of ylide would be largely increased [7]. Accordingly, the extra stability required developing appropriate reaction conditions to activate sulfur ylides for being as reactants. Recent reports have shown promise that stabilized sulfur ylides could be employed as fluoroalkylation reagents.
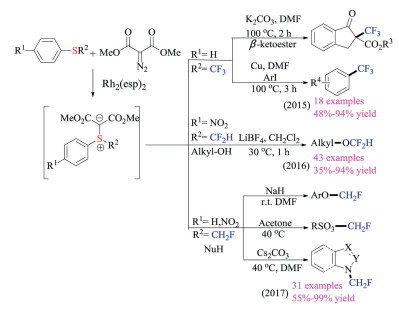
Shen group developed a concise Rh-catalyzed carbenoid addition strategy to synthesize a series of fluorine-containing sulfonium ylides from corresponding thioethers (Scheme 15) [28]. These easily available reagents were not sensitive to either air or moisture, and could serve as electrophilic trifluoromethylation, difluoromethylation and monofluoro-methylation reagents. As shown in Scheme 15, a great variety of nucleophiles were amenable to give fluoroalkylated products in good yields under mild reaction conditions.

|
Download:
|
| Scheme 15. Shen's work on using sulfur ylide as fluoroalkylation reagents. | |
{kind=link}
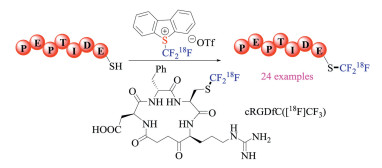
Compared to sulfur ylides, sulfonium salts had a long history of being employed as reagents to incorporate fluorine-containing groups. The combination of "soft" sulfur and "hard" fluorine in these reagents along with the unique reactivity of sulfur-containing compounds have rendered them ideal for being potential fluorination reagents [29]. Recently, such reagents have also been increasingly utilized in radio-labeling. In 2018, Gouverneur et al. synthesized the 18F-labeled Umemoto reagents by a fluoride exchange and oxidative cyclization cascade (Scheme 16) [30]. The resulting reagents could be employed for direct 18F-labeling of unmodified peptides at the cysteine residue via S-trifluoromethylation. Biodistribution studies further confirmed the 18F-SCF3 moiety was viable for imaging purposes.

|
Download:
|
| Scheme 16. 18F-labeling of unmodified peptides. | |
{kind=link}
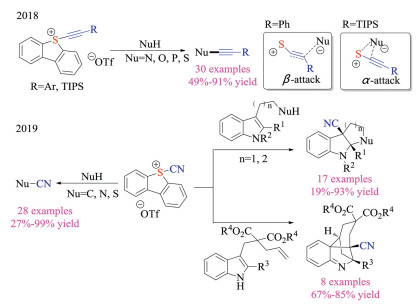
Moreover, sulfonium salts could have been employed as electrophilic reagents for other types of bond formations. Alcarazo and co-workers have recently synthesized a series of (alkynyl)dibenzothiophenium triflates and demonstrated their potential application as electrophilic sulfur-based alkynylation reagents (Scheme 17) [31a]. Various carbon, nitrogen, sulfur, and phosphine nucleophiles were amenable to afford desired alkynylation products in good yields, attesting to the geniality and generality of the protocol. Interestingly, the reagents displayed different reactive sites toward nucleophilic substitutions, depending on the terminal substitution group ("Si" vs. Ar). Later, the same group employed a similar synthetic pathway to prepare the 5-(cyano)dibenzothiophenium triflates [31b]. These novel sulfonium salts displayed distinctive reactivities, as it not only reacted with a number of nucleophiles but also participated in Friedel-Craft type reactions under Lewis acid-free conditions. Exceptionally, this new reagent was applicable for electrophilic cyanation-Povarov cascade, affording the tetracyclic products in good efficiency.

|
Download:
|
| Scheme 17. Sulfonium salts as alkynylation and cyanation reagents. | |
{kind=link}
3. As reaction intermediates
Both sulfonium salt and sulfur ylides had a rich and fruitful history in being employed as key intermediate for synthesizing organosulfur compounds. In this section, we only focused on two strategies utilizing either carbene or aryne/alkyne chemistry to generate such essential sulfur-containing intermediates.
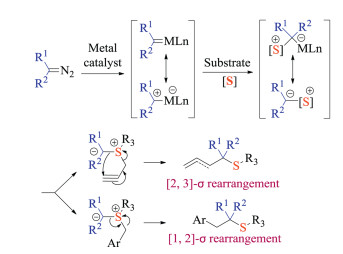
3.1. Carbene induced reactionsTransition metals could enable the decomposition of diazo compounds to generate metal carbene species in situ [32]. As electrophilic species, they readily reacted with thioethers to form corresponding sulfonium ylide intermediates. Both the [2,3]-sigmatropic and [1,2]-sigmatropic rearrangement products could be selectively liberated by simply varying the substitution group patterns (Scheme 18). Important advances have arisen from both developing new catalysts and expanding reaction scope. As such, this part mainly covered recent efforts on asymmetric catalysis as well as transformations under green conditions.

|
Download:
|
| Scheme 18. Carbene induced sulfonium and sulfur ylide intermediate formation. | |
{kind=link}
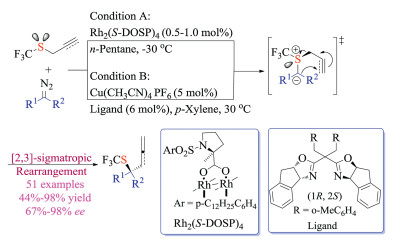
Wang group had long-standing interests in metal carbene chemistry and their applications in sulfur ylides based transformations [33]. In 2017, Wang and co-workers reported the first example of catalytic enantioselective [2,3]-sigmatropic rearrangement (Doyle-Kirmse) reaction of allyl trifluoromethyl sulfides (Scheme 19). This cutting edge protocol employed easily available chiral Rh(Ⅱ) or Cu(Ⅰ) catalyst, performed under mild conditions, and demonstrated great substrate scope as well as functional group tolerance (51 examples, up to 98% yield and 98% ee) [33c]. Significantly, this reaction offered a new protocol for constructing quaternary stereogenic centers bearing a trifluoromethylthio group (-SCF3). Mechanistic studies suggested a free ylide intermediate rather than the metal-bound ylide. The chiral carbene could differentiate the heterotopic lone-pair of sulfur atom to form relatively stable chiral sulfur ylide, and the observed enantioselectivity was realized through subsequent transfer of the chirality from sulfur to carbon via concerted rearrangements.

|
Download:
|
| Scheme 19. Wang's enantioselective [2,3]-sigmatropic rearrangement. | |
{kind=link}
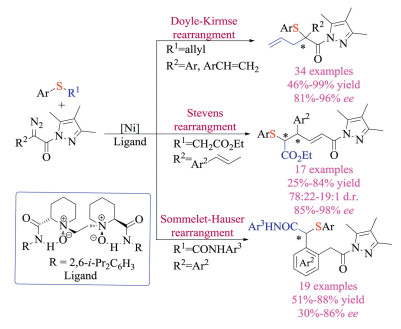
In 2018, Feng and co-workers achieved the Ni-catalyzed highly enantioselective Doyle-Kirmse reaction of α-diazo pyrazole amides by using their privileged chiral N, N-dioxide ligands (Scheme 20) [34a, b]. The protocol was amenable to a wide range of α-diazo pyrazoleamides and sulfides, giving rearrangement products with high yields and enantioselectivities (up to 99% yield and 96% ee). The choice of pyrazolyl group proved to be crucial. Firstly, it facilitated the formation of metal carbene with assistance of Ni complex. Secondly, it transferred either free ylides or metal-bonded ylides to chiral Lewis-acid-bonded ylide via strong bidentate coordination with chiral catalysts, and thus provided excellent enantioselection. Thirdly, it served as latent synthetic handle that subsequent functional group transformation could easily afford chiral alcohol, amide and ester derivatives without compromising of ee. Later, Feng and co-workers also developed the asymmetric [2, 3] Stevens and Sommelet-Hauser rearrangements of sulfonium ylides using either vinyl- or aryl-substituted α-diazo pyrazoleamides under similar conditions [34c]. Again, control experiments indicated the necessity of pyrazolyl groups for both reaction reactivity and stereocontrol. However, unlike the early report, mechanistic studies revealed that the chiral catalyst could discriminate between the heterotopic lone pairs of sulfur in substrates, and govern both the 1, 3-proton transfer as well as subsequent rearrangements in an enantioselective fashion. Interestingly, deuterium-labelling experiments revealed that the intermolecular 1, 3-proton transfer was assisted by the carboxylic acid additives.

|
Download:
|
| Scheme 20. Feng's asymmetric rearrangement reactions. | |
{kind=link}
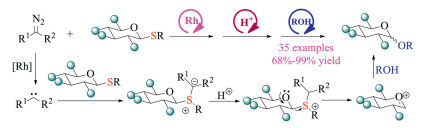
Recently, Wan and co-workers developed a highly efficient glycosylation protocol using sequential rhodium and Brønsted acid catalysis (Scheme 21) [35]. Initially, the rhodium-catalysis enabled the formation of carbenes and resulting sulfonium ylides. Next, the Brønsted acids promoted the formation of sulfonium salt intermediates, which served as a leaving group for further glycosylation. This well-designed sequential catalysis indeed offered a new powerful tool for activating thioglycosides, delivering a variety of glycosyl donors in good yields. The reported protocol also exhibited low catalyst loading and mild reaction conditions, which should be of great utilities in the carbohydrate chemistry and related natural products synthesis.

|
Download:
|
| Scheme 21. Thioglycosides activation via tandem rhodium and Brønsted acid catalysis. | |
{kind=link}
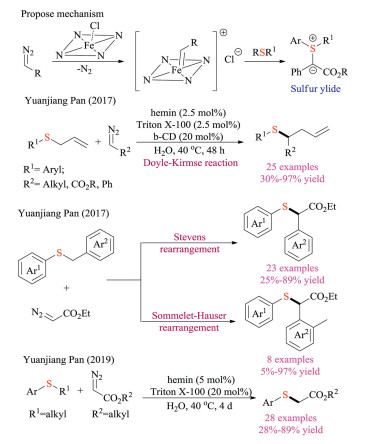
Pan and co-workers recently established a hemin-based catalytic system to form metal carbenoids at aqueous conditions (Scheme 22) [36]. In 2017, they first reported its application to the Doyle-Kirmse reaction [36a]. Both β-CD and Triton X-100 were utilized as additives to facilitate the reaction synergistically. The proposed mechanism similarly involved the formation of sulfonium ylide through an iron-carbenoid species, followed by classic [2,3]-sigmatropic rearrangement to afford the desired product in good yields. This protocol also showed broad substrate scope under mild conditions and possessed relatively lower E-factor owing to using water as the solvent. Interestingly, allyl amine and allyl ether analogous proved to be unamenable under standard conditions. Later, they also reported the hemin catalyzed competing [1,2]-Stevens and Sommelet-Hauser rearrangements of benzyl sulfonium ylides [36b]. A series of substrates and solvents were screened to explore the chemoselectivity. Experiment results indicated that the benzyl group bearing an electron-withdrawing group was prior to [2,3]-Sommelet-Hauser rearrangement, whereas electron-donating groups made [1,2]-Stevens rearrangement overweigh. Meanwhile, water was also discovered to promote the Sommelet–Hauser pathway. Very recently, the catalytic dealkylation of sulfonium salts was also developed by Pan and co-workers using the same Hemin system in aqueous solvents [36c].

|
Download:
|
| Scheme 22. Reaction of carbene and sulfides under aqueous conditions. | |
{kind=link}
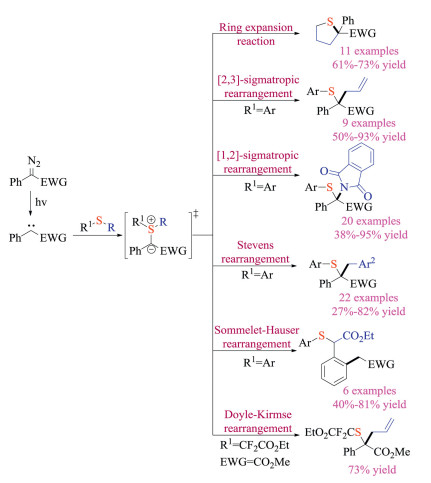
Koenigs and co-workers recently demonstrated that the blue LEDs could enable the photochemical generation of carbene species from diazoalkanes (Scheme 23) [37a]. This transition-metal-free and operationally simple paradigm, therefore offered an alternatively efficient pathway to generate sulfur ylide intermediates. Starting from 2018, Koenigs group has successively developed a series of transformations on carbene and sulfide under such photochemical conditions, as shown in Scheme 23 [37]. Moreover, they achieved the photochemical Doyle-Kirmse reaction under continuous-flow conditions using a simple glass micro-reactor. Obviously, this operationally simple approach avoided using Rh catalysts, and should open new possibilities for modern carbene chemistry and set base for potential applications in both academic and industrial settings.

|
Download:
|
| Scheme 23. Koenigs' systematic work on photochemical generation of carbenes. | |
{kind=link}
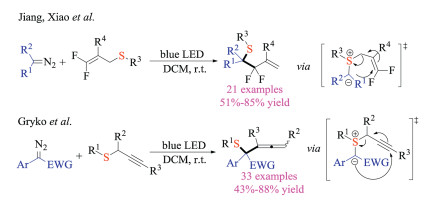
Since these seminal reports, there have been many studies that expanded upon this strategy for synthesizing organosulfur compounds. In 2019, Xiao, Jiang and co-workers developed a blue light mediated formal Doyle-Kirmse reaction of difluoroallyl sulfides (Scheme 24) [38]. This protocol displayed good substrate scope, providing quick access to highly functionalized gem-difluoroallyl containing esters. Later in the same year, Gryko et al. reported the photochemical Doyle-Kirmse reaction of propargyl sulfides with diazo compounds [39]. A wide array of densely functionalized allene products were obtained in good to excellent yield conveniently.

|
Download:
|
| Scheme 24. Recent advances in Doyle-Kirmse reaction under photochemical conditions. | |
{kind=link}
3.2. Aryne/alkyne induced reactions
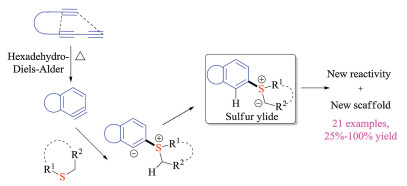
Arynes with lower-lying LUMO could react with various nucleophiles to build molecular complexity with high efficacy. It has recently drawn great attention as an alternative approach to prepare S(Ⅳ) intermediates via nucleophilic addition [40]. Hoye group has long-term interests in developing hexadehydro-Diels-Alder (HDDA) based transformations [41]. In 2016, they investigated the reaction between HDDA-generated benzyne derivatives and various sulfides, providing insights into both mechanism and reactivities (Scheme 25) [41a]. The aryne intermediates were in-situ formed via the intramolecular thermal cycloaddition of tethered triynes. The nucleophilic addition of thioethers to arynes subsequently formed the 1, 3-zwitterionic intermediates. Upon proton transfer, they could undertake either sigmatropic rearrangement reactions or three-component ring-opening reactions. Indeed, this protocol offered a new pathway to access sulfonium salt of sulfur ylide intermediates with unique scaffolds.

|
Download:
|
| Scheme 25. HDDA reaction of sulfides. | |
{kind=link}
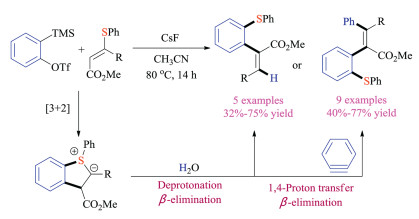
Later in the same year, another breakthrough was reported by Studer's group. They developed a unique reaction between aryne and vinyl sulfides, introducing a new strategy for formation of sulfonium ylides (Scheme 26) [42]. Experimental studies and DFT calculations indicated that the electrophilic aryne species initially reacted with vinyl thioethers via an unprecedented [3 + 2] cycloaddition pathway to form the annulated sulfur ylide intermediates, which could either be protonated by water or reacted with another equivalent of aryne to synthesize a large variety of di-, tri- and tetra-substituted alkenes in good efficiency.

|
Download:
|
| Scheme 26. Reaction of aryne with vinyl sulfides. | |
{kind=link}
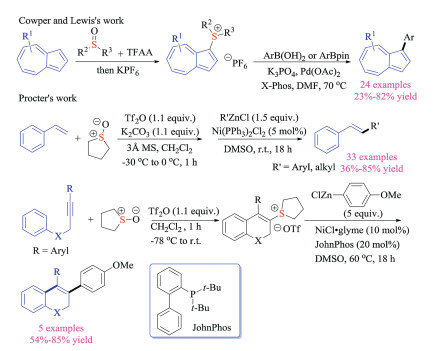
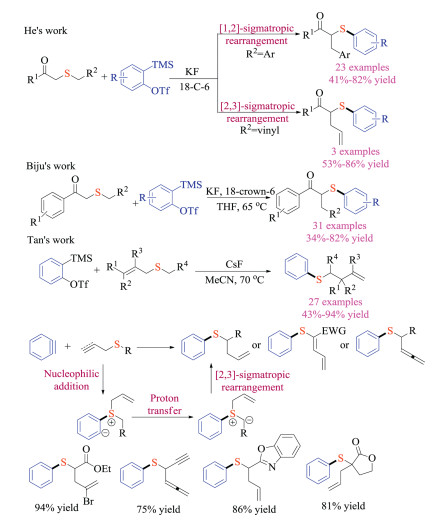
Such ground-breaking works have given rise to the rapidly developing domain of aryne and organosulfur chemistry. In 2017, He, Biju and Tan group independently reported the aryne induced rearrangement reaction of sulfur ylides (Scheme 27) [43]. He and co-workers first discovered that simple β-keto thioethers with different substitution patterns underwent either [1,2]- or [2,3]-sigmatropic rearrangement reaction with arynes, providing quick access to β-keto thioether compounds [43a]. In the meantime, Biju and co-worker focused on the [2,3]-Stevens rearrangement reaction of allyl thioethers, which could be applicable to construct quaternary carbon centers [43b]. In comparison with Biju and He's reports, the [2,3]-sigmatropic rearrangement developed by Tan et al. tolerated a wide range of electron-withdrawing groups that were used to facilitate the formation of sulfur ylides [43c]. Propargylic thioethers were also viable substrates to give allene or diene products in good yields. Again, the nucleophilic addition of thioethers to arynes generated key zwitterionic intermediate that underwent proton transfer to provide the key sulfur ylides.

|
Download:
|
| Scheme 27. aryne induced sigmatropic rearrangement of thioethers. | |
{kind=link}
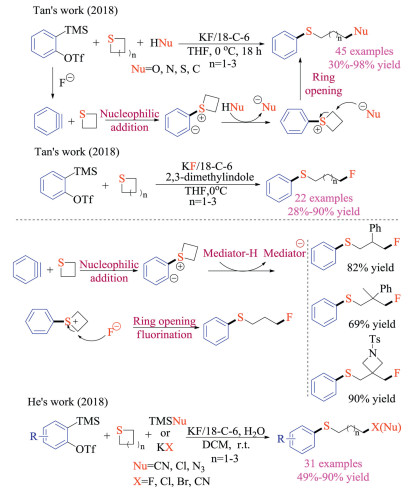
Tan and co-workers also successfully reported the three-component reaction of Kobayashi aryne precursors, cyclic sulfides, and nucleophiles, giving structurally diverse thioethers in good yields (Scheme 28) [44]. Thietanes could be activated via the addition to electrophilic arynes, which were formed under basic fluoride conditions. The zwitterion species could then deprotonate pro-nucleophiles, and they would consecutively attack the activated carbon-sulfur bond on cyclic sulfides to complete the ring-openings. This transition metal-free reaction proceeded under mild reaction conditions and displayed exceptional generality of nucleophiles (C, O, S and N centered nucleophiles), offering a complementary method to Hoyes' HDDA chemistry [41].

|
Download:
|
| Scheme 28. Aryne induced ring opening reactions of cyclic sulfides. | |
{kind=link}
Based on these results, the same group then developed the aryne induced ring-opening fluorination reaction of cyclic thioethers, using readily available potassium fluoride salts as a fluoride source [45a]. They successfully identified 2, 3-dimethylindole as a suitable mediator. In addition to being a proton donor to capture ylides to form sulfonium intermediate, the deprotonated 2, 3-dimethylindole could neither react with arynes, nor enable the competing ring-opening reaction as a nucleophile. Given the significant role of fluorine atoms in medicinal and material chemistry, this protocol should be of great synthetic value. Noteworthy, He and co-worker simultaneously reported a multicomponent reaction of benzynes, cyclic thioethers and non-proton containing nucleophiles [45b]. In the presence of water as the mediator, both potassium salts and trimethylsilyl reagents could be employed to achieve the ring-opening reaction in moderate to good yields.
More recently, Meng and co-workers developed a novel domino reaction of thioaurone analogues and arynes, synthesizing a large variety of tricyclic eight-membered and [5,5]-bicyclic sulfides (Scheme 29) [46]. The aryne first underwent a [3 + 2] cycloaddition reaction with the vinyl sulfide fragment to generate the 1, 2-dipole species in situ, subsequent proton transfer/C–S bond cleavage eventually gave the final product. The tentative mechanism was further proved by DFT calculations and control experiments.

|
Download:
|
| Scheme 29. Reaction of arynes with thioaurone analogues. | |
{kind=link}
Reactions of aryne with sulfoxides proceeded with different mechanism (Scheme 30). In 2014, Xiao group reported the pioneering work on three-component reactions of aryne, DMSO and α-bromo carbonyl compounds [47a]. The title reaction initiated with the cycloaddition of aryne with S=O motif to form the four-membered ring intermediate, which upon subsequent cleavage of the S-O bond led to zwitterionic sulfonium salt intermediates. Based on the reported reaction pathway, many groups then independently designed and developed new transformations on arynes and sulfoxides. Wang group reported the direct oxythiolation reaction of arynes, utilizing either dimethyl sulfoxides or methyl aryl sulfoxides. The key 1, 4-zwitterionic sulfonium intermediate reacted with another molecule of aryne species, and concomitant dealkylation provided the O-arylthio-substituted diaryl ether product in moderate yields. Gogoi et al. then reported a similar three-component cascade of aryne, DMSO, and activated alkyne, whereas the phenolate anion was trapped with acetylenedicarboxylate.

|
Download:
|
| Scheme 30. Reactions of aryne with sulfoxides. | |
{kind=link}
Moreover, by varying the substitution groups on sulfoxides, new reactivities have been discovered. Li and co-workers developed a well-designed aryne 1, 2, 3-trifunctionalization reaction using Kobayashi aryne precursor with aryl allyl sulfoxides. The reaction proceeds with the formal [2 + 2] cycloaddition, S to O allyl migration, and subsequent Claisen rearrangement. Studer and co-workers reported the reaction of arynes with aryl vinyl sulfoxides. In this case, the reaction proceeded with tandem S-O bond cleavage of the four-membered adduct and ionic vinyl group migration, which provided quick access to di- or tri-substituted vinyl ethers with excellent stereospecificity.
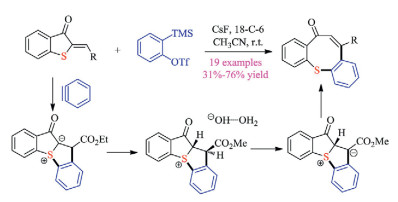
Recently, Hosoya group discovered that if the reaction of aryne and diarylsulfoxide was conducted at high temperature (80-110 ℃), the sulfonium based 1, 4-zwitterionic intermediate proceed with migratory O-arylation to give a wide array of O-arylthio-substituted diarylethers in moderate to good yields. In comparison, Peng and co-workers revealed that simply treating benzyne with diarylsulfoxides at room temperature could furnish the O-aryloxy triarylsulfonium salt products. A sequential [2 + 2] cycloaddition/O-arylation/protonation mechanism is proposed. Interestingly, Peng et al. later discovered that by slightly modifying the reaction conditions, the arynes could function as "reductants" to convert diarylsulfoxides and heteroaryl sulfoxides to biaryls and desulfurized heteroarenes, respectively. Further investigation suggested that diarylsulfoxides bearing electro-donating groups were not amenable substrates, which demonstrated certain limitations of this protocol. Mechanistic studies and DFT calculation suggested that a tetraaryl(heteroaryl) sulfuranes intermediate and corresponding π-complex with Cs+ might be responsible for the reported unique reactivities.
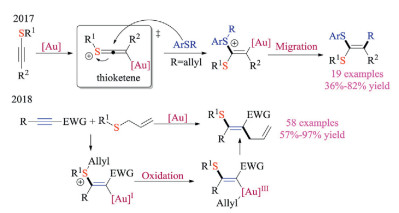
Upon activation with corresponding catalysts, less reactive alkynes could also form the sulfonium intermediate via nucleophilic addition of thioethers (Scheme 31). In 2017, Shi and co-workers reported the gold-catalyzed nucleophilic addition reactions of thioalkynes [48a]. Based on the observed cis-addition resulted from a gold-associated thioketene intermediate, the addition of thioethers to thioalkyne reaction was developed. The migration reaction of R groups from the in-situ formed sulfonium intermediates gave a variety of ketene dithioacetal products in good yields. The gold catalyst, although poisoned by sulfur, clearly altered the reactivity of thioalkyne toward nucleophilic additions. Based on these findings, they later reported a novel gold redox catalysis protocol for stereoselective thioallylation of alkynes [48b]. The sulfonium intermediates were formed by the addition of allylic thioethers to electron-deficient alkynes under Au catalysis. More importantly, the sulfonium intermediates, as the author proposed, could assist the oxidation of Au(Ⅰ) to Au(Ⅲ). The tentative mechanism was further confirmed by both mass spectrum studies and control experiments.

|
Download:
|
| Scheme 31. Shi's work on thioalkyne activation via S(Ⅳ) intermediates. | |
{kind=link}
Another notable example was the scandium-catalyzed site-selective and stereoselective thio-allylation reaction of allenyl imidates reported by Bandini and co-workers in 2019 (Scheme 32) [49]. The protocol allowed for synthesizing β-thio, γ-allyl-α, β-unsaturated carboxylic derivatives in good efficiencies. The loading of Scandium(Ⅲ) triflates could be reduced to 3 mol% when the reaction was scaled up to grams. Detailed DFT calculation studies suggested a plausible cascade conjugate addition/[3,3]-sigmatropic rearrangement pathway, and rationalized the observed high selectivities in the meanwhile.

|
Download:
|
| Scheme 32. Tandem Michael addition/sigmatropic rearrangement reaction. | |
{kind=link}
3.3. Other breakthroughs
Inspired by the progress of sulfur ylide chemistry, Huang et al. recently reported the first example that organic sulfide catalyst could activate both benzyl halides and arylboronic acids to achieve C(sp3)-C(sp2) Suzuki-Miyaura cross-coupling reaction (Scheme 33) [50]. Various diarylmethane products were obtained in good yields under transition-metal-free conditions. This protocol started with the nucleophilic substitution reaction of sulfide catalysts with benzylic halides, proceeded through both sulfonium and sulfur ylide intermediates. Subsequent zwitterionic boron "ate" complex formation, 1, 2-metallate shift along with protodeboronation gave the overall coupling products. Compared to aryl bromides and aryl iodides which are reactive in classical transition metal-catalyzed coupling reactions, the exclusive selectivity of benzyl chlorides observed in this protocol should offer great synthetic flexibility in design and synthesis complex molecules.

|
Download:
|
| Scheme 33. Sulfide catalyzed Suzuki-Miyaura coupling reaction. | |
{kind=link}
Developing methods for chemically modifying proteins offered great opportunities to attach functional scaffolds such as fluorescent dyes and small-molecule drugs to the proteins. Gaunt and co-workers have recently developed a well-tuned hypervalent iodine reagent for fast protein functionalization via formation of sulfoniums (Scheme 34) [51]. Instead of targeting cysteine, tyrosine or lysine residue, this approach enabled chemoselectively labelling of S-Me group on the side chain of methionine residues under aqueous conditions, and could be achieved at low-micromolar concentrations. Additives including thiourea, formic acid and TEMPO were found to be indispensable in facilitating the target transformation as well as inhibiting the potential over-oxidation and decomposition. More importantly, the obtained protein conjugates were relatively stable, yet highly reactive toward further radical coupling reactions under photochemical conditions. In this case, the combination of these two transformations could serve as a platform approach for protein functionalization, which should inspire scientists to explore new applications and demonstrate unforeseen possibilities.

|
Download:
|
| Scheme 34. Methionine-selective bio-conjugation. | |
{kind=link}
4. Catalysts
Ammonium salts and phosphonium salts have been widely employed as organocatalysts in both academic and industrial settings [52]. Sulfonium salts with strong acidic α-hydrogens, however, have seldom been reported as catalysts, owing especially to the high reactivity and low stability of these compounds under basic conditions.
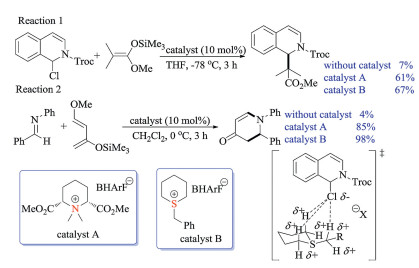
Shirakawa group recently employed trialkylsulfonium salts as hydrogen bonding catalysts, making the best use of the strong acidity of the α-hydrogen atoms (Scheme 35) [53]. The catalytic ability was demonstrated by the Mannich, aza Diels–Alder and imine reduction reactions, which showed superior reactivities over corresponding tetraalkylammonium salts. Furthermore, both X-ray crystal structures and the NMR titration experiments for binding abilities provided solid evidence that these sulfonium salts activated chloride substrates via hydrogen bonding interactions.

|
Download:
|
| Scheme 35. Trialkyl sulfonium salts as hydrogen bonding catalysts. | |
{kind=link}
Asymmetric phase transfer catalysis has been widely recognized as a green and sustainable approach, and has evolved into one of the most practical asymmetric synthesis methods [54]. Previous efforts on asymmetric phase transfer catalysis mainly focused on using chiral ammonium salts as catalysts. Based on above-mentioned results, Shirakawa and coworkers in 2018 reported the first example of chiral bifunctional trialkylsulfonium salts catalyzed highly enantioselective conjugate addition reaction of 3-substituted oxindoles to maleimides (Scheme 36) [55]. Both sulfonium and urea motifs were found to be essential for obtained good yields and enantioselectivities. The well-designed reaction performed under base free neutral phase transfer conditions, avoiding the weakness of sulfonium salts. In this case, the sulfonium motif on the catalyst should function via ionic interaction. These creative reports should open a new dimension of sulfonium salt chemistry in organic synthesis.

|
Download:
|
| Scheme 36. Chiral sulfonium salts catalyzed asymmetric phase transfer Michael addition reaction. | |
{kind=link}
5. Summary and outlook
The past several years have witnessed reviving interests from the community in the chemistry of sulfonium salt and sulfur ylide. The overview of related research reports ranging from 2016 to 2019 was divided into three sections as shown above. To aid the readers of Chinses Chemical Letters, transformations on the same topics are organized together in each of section. As either reactants or intermediates, both sulfonium salts and sulfur ylides have displayed unique reactivities in a wide range of transformations to provide quick and often selective access to organosulfur compounds in great structural diversities. The possible reaction mechanism is also illustrated in detail for most of notable examples. Moreover, many efforts have also been made for match growing green and sustainable requirements. Interdisciplinary applications of such chemistry in radio-labelling and bioconjugation have also shown great promise. Significantly, the advantages and limitations of those works were emphasized, compared, and critically discussed.
Taken together, "new tricks" have been developed for sulfonium salt and sulfur ylide chemistry, despite their fruitful history. Looking forward, we see the following challenges aka opportunities that might drastically change the field in the upcoming decade, year, or even month. First, despite the level of pursuit in the first two sections, what has been underdeveloped to date is the use of sulfonium salts as organocatalysts. How to make the best use of sulfonium salts while avoiding their weakness is crucial for enabling catalysis. Further developments to fill this apparent technology gap are highly expected. Besides, expanding the reaction scope and discovering new reactivities beyond the state-of-the-art approaches remains to be an elusive task. For instance, even though the sulfonium salts are increasingly utilized in organic photochemistry, their applications in electrosynthesis haven't reach full potentials [56]. Another shortcut for chemists to reach innovative discoveries would be getting inspiration and learning from nature. For instance, S-Adenosyl-L-methionine (SAM) is an enzyme bearing sulfonium salt motif, which can participate in both ionic and radical type reactions. Mimicking its systematic bond-formation and breakage principle should receive particular attentions in this regard. Last but not least, the multidisciplinary effort needs to be put into context to bring another leap forward. The stereotype of research that lack of interdisciplinary applications in academia is gradually outdated. Integration of sulfonium salt and sulfur ylide chemistry into other rapid developing domains, such as material chemistry and biochemistry, is a must for future research. As such, numerous work remains to unlock full potentials of such S(Ⅳ) species, and future success must rely mainly on improvement of green and sustainable methods, broadening of reaction scopes, transferring into the multidisciplinary applications, and de novo development of nature-inspired reactivities. We strongly hope that this concise review will serve as a starting point for scientists looking to enter this exciting field and provide inspiration for future discoveries.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsJ. Tan is grateful for the support from the National Natural Science Foundation of China (No. 21702013), and the Fundamental Research Funds for the Central Universities (Nos. XK1802-6 and buctrc201721) at the BUCT. J. Tan and Y. Liu are thankful for the support from the Open Project Program (No. SPFW2019YB06) of Beijing Key Laboratory of Flavor Chemistry, Beijing Technology and Business University (BTBU).
| [1] |
(a) R.J. Cremlyn, An Introduction to Organosulfur Chemistry, John Wiley & Sons, Hoboken, 1996; (b) P.C.B. Page, Organosulfur Chemistry I., Springer, Heidelberg, 1999; (c) C.M. Rayner, Advances in Sulfur Chemistry, JAI Press, Greenwich, 2000; (d) A.Q. Acton, Sulfur Compounds: Advances in Research and Application, Scholarly Editions, Atlanta, 2012. |
| [2] |
(a) E.A. Ilardi, E. Vitaku, J.T. Njardarson, J. Med. Chem. 57(2014) 2832-2842; (b) B.R. Smith, C.M. Eastman, J.T. Njardarson, J. Med. Chem. 57(2014) 9764-9773. |
| [3] |
(a) K.L. Dunbar, D.H. Scharf, A. Litomska, C. Hertweck, Chem. Rev. 117(2017) 5521-5577; (b) M.H. Feng, B.Q. Tang, S. Liang, X.F. Jiang, Curr. Top. Med. Chem. 16(2016) 1200-1216. |
| [4] |
(a) L.D. Wang, W. He, Z.K. Yu, Chem. Soc. Rev. 42(2013) 599-621; (b) S.G. Modha, V.P. Mehta, E.V. Van der Eycken, Chem. Soc. Rev. 42(2013) 5042-5055; (c) D. Kaiser, I. Klose, R. Oost, et al., Chem. Rev. 119(2019) 8701-8780. |
| [5] |
(a) A.W. Johnson, R.B. Lacount, J. Am. Chem. Soc. 83(1961) 417-423; (b) E.J. Corey, M. Chaykovsky, J. Am. Chem. Soc. 84(1962) 867-868; (c) S.Q. Guo, N.N. Zhang, X.Z. Tang, et al., Chin. Chem. Lett. 30(2019) 406-408. |
| [6] |
L. Kurti, B. Czako, Strategic Applications of Named Reactions in Organic Synthesis. Burlington: Elsevier, 2005: pp. 294-295.
|
| [7] |
(a) R. Oost, R.J.D. Neuhaus, J. Merad, N. Maulide, Sulfur Ylides in Organic Synthesis and Transition Metal Catalysis, Structure and Bonding, Springer, Berlin, Heidelberg, 2017; (b) J.D. Neuhaus, R. Oost, J. Merad, N. Maulide, Top. Curr. Chem. 376(2018) 15; (c) L.Q. Lu, T.R. Li, Q. Wang, W.J. Xiao, Chem. Soc. Rev. 46(2017) 4135-4149; (d) M. Mondal, S. Chen, N.J. Kerrigan, Molecules 23(2018) 738-767; (e) F. Pan, Z.J. Shi, ACS Catal. 4(2014) 280-288; (f) K. Gao, S. Otsuka, A. Baralle, et al., J. Synth. Org. Chem. Jpn. 74(2016) 1119-1127; (g) D.H. Ortgies, A. Hassanpour, F. Chen, et al., Eur. J. Org. Chem. 2016(2016) 408-425; (h) S. Otsuka, K. Nogi, H. Yorimitsu, Top. Curr. Chem. 376(2018) 13; (i) P. Chauhan, S. Mahajan, D. Enders, Chem. Rev. 114(2014) 8807-8864. |
| [8] |
(a) T.J. Colacot, New Trends in Cross-Coupling: Theory and Applications, 4th. ed., Royal Society of Chemistry, British, 2014; (b) Á. Molnár, Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments, Wiley-VCH, Weinheim, Germany, 2013; (c) M.L. Crawley, B.M. Trost, Applications of Transition Metal Catalysis in Drug Discovery and Development, Wiley, New York, 2012; (d) J.J. Tan, Y.G. Chen, H.M. Li, N. Yasuda, J. Org. Chem. 79(2014) 8871-8876. |
| [9] |
J. Srogl, G.D. Allred, L.S. Liebeskind, J. Am. Chem. Soc. 119 (1997) 12376-12377. DOI:10.1021/ja9726926 |
| [10] |
(a) Z.Y. Tian, Y.T. Hu, H.B. Teng, C.P. Zhang, Tetrahedron 59(2018) 299-309; (b) S.M. Wang, H.X. Song, X.Y. Wang, et al., Chem. Commun. (Camb. ) 52(2016) 11893-11896; (c) S.M. Wang, X.Y. Wang, H.L. Qin, C.P. Zhang, Chem. Eur. J. 22(2016) 6542-6546; (d) Z.Y. Tian, S.M. Wang, S.J. Jia, et al., Org. Lett. 19(2017) 5454-5457; (e) Z.Y. Tian, C.P. Zhang, Chem. Commun. (Camb. ) 55(2019) 11936-11939; (f) X.Y. Wang, H.X. Song, S.M. Wang, et al., Tetrahedron 72(2016) 7606-7612. |
| [11] |
(a) D. Uno, H. Minami, S. Otsuka, et al., Chem. Asian J. 13(2018) 2397-2400; (b) H. Minami, K. Nogi, H. Yorimitsu, Org. Lett. 21(2019) 2518-2522; (c) H. Minami, S. Otsuka, K. Nogi, H. Yorimitsu, ACS Catal. 8(2018) 579-583. |
| [12] |
D.C. Simkó, P. Elekes, V. Pázmándi, Z. Novák, Org. Lett. 20 (2018) 676-679. DOI:10.1021/acs.orglett.7b03813 |
| [13] |
P. Cowper, Y. Jin, M.D. Turton, et al., Angew. Chem. Int. Ed. 55 (2016) 2564-2568. DOI:10.1002/anie.201510666 |
| [14] |
M.H. Aukland, F.J.T. Talbot, J.A. Fernández-Salas, et al., Angew. Chem. Int. Ed. 57 (2018) 9785-9789. DOI:10.1002/anie.201805396 |
| [15] |
(a) D. Ravelli, S. Protti, M. Fagnoni, Chem. Rev. 116(2016) 9850-9913; (b) Q.Q. Zhou, Y.Q. Zou, L.Q. Lu, W.J. Xiao, Angew. Chem. Int. Ed. 58(2019) 1586-1604; (c) N.A. Romero, D.A. Nicewicz, Chem. Rev. 116(2016) 10075-10166. |
| [16] |
I. Ghosh, L. Marzo, A. Das, et al., Acc. Chem. Res. 49 (2016) 1566-1577. DOI:10.1021/acs.accounts.6b00229 |
| [17] |
S. Donck, A. Baroudi, L. Fensterbank, et al., Adv. Synth. Catal. 355 (2013) 1477-1482. DOI:10.1002/adsc.201300040 |
| [18] |
S. Otsuka, K. Nogi, T. Rovis, H. Yorimitsu, Chem. Asian J. 14 (2019) 532-536. DOI:10.1002/asia.201801732 |
| [19] |
(a) F. Berger, M.B. Plutschack, J. Riegger, et al., Nature 567(2019) 223-228; (b) J.K. Li, J.T. Chen, R.C. Sang, et al., Nat. Chem. 12(2020) 56-62; (c) P.S. Engl, A.P. Häring, F. Berger, et al., J. Am. Chem. Soc. 141(2019) 13346-13351; (d) F. Ye, F. Berger, H. Jia, et al., Angew. Chem. Int. Ed. 58(2019) 14615-14619; (e) R.C. Sang, S.E. Korkis, W.Q. Su, et al., Angew. Chem. Int. Ed. 58(2019) 16161-16166. |
| [20] |
C. Huang, J. Feng, R. Ma, et al., Org. Lett. 21 (2019) 9688-9692. DOI:10.1021/acs.orglett.9b03850 |
| [21] |
S. Rohrbach, A.J. Smith, J.H. Pang, et al., Angew. Chem. Int. Ed. 58 (2019) 16368-16388. DOI:10.1002/anie.201902216 |
| [22] |
(a) V. Bernard-Gauthier, T.L. Collier, S.H. Liang, N. Vasdev, Drug Discov. Today Technol. 25(2017) 19-26; (b) G. Pascali, L. Matesic, B. Zhang, et al., EJNMMI Radiopharm. Chem. 2(2017) 9-26. |
| [23] |
(a) L.J. Mu, C.R. Fischer, J.P. Holland, et al., Eur. J. Org. Chem. 2012(2012) 889-892; (b) K. Sander, T. Gendron, E. Yiannaki, et al., Sci. Rep. 5(2015) 9941-9945. |
| [24] |
T. Gendron, K. Sander, K. Cybulska, et al., J. Am. Chem. Soc. 140 (2018) 11125-11132. DOI:10.1021/jacs.8b06730 |
| [25] |
P. Xu, D. Zhao, F. Berger, et al., Angew. Chem. Int. Ed. 59 (2020) 1956-1960. DOI:10.1002/anie.201912567 |
| [26] |
(a) X.X. Ming, Z.Y. Tian, C.P. Zhang, Chem. Asian J. 14(2019) 3370-3379; (b) Z.Y. Tian, X.X. Ming, H.B. Teng, et al., Chem. Eur. J. 24(2018) 13744-13748. |
| [27] |
J.N. Zhao, M. Kayumov, D.Y. Wang, A. Zhang, Org. Lett. 21 (2019) 7303-7306. DOI:10.1021/acs.orglett.9b02584 |
| [28] |
(a) Y.F. Liu, X.X. Shao, P.P. Zhang, et al., Org. Lett. 17(2015) 2752-2755; (b) J.S. Zhu, Y.F. Liu, Q.L. Shen, Angew. Chem. Int. Ed. 55(2016) 9050-9054; (c) Y.F. Liu, L. Lu, Q.L. Shen, Angew. Chem. Int. Ed. 56(2017) 9930-9934. |
| [29] |
(a) C.F. Ni, M.Y. Hu, J.B. Hu, Chem. Rev. 115(2015) 765-825; (b) C. Zhang, Org. Biomol. Chem. 12(2014) 6580-6589; (c) S. Barata-Vallejo, B. Lantaño, A. Postigo, Chem. Eur. J. 20(2014) 16806-16829; (d) G.K. Liu, X. Li, W.B. Qin, et al., Chin. Chem. Lett. 30(2019) 1515-1518. |
| [30] |
S. Verhoog, C.W. Kee, Y.L. Wang, et al., J. Am. Chem. Soc. 140 (2018) 1572-1575. DOI:10.1021/jacs.7b10227 |
| [31] |
(a) B. Waldecker, F. Kraft, C. Golz, M. Alcarazo, Angew. Chem. Int. Ed. 57(2018) 12538-12542; (b) X.D. Li, C. Golz, M. Alcarazo, Angew. Chem. Int. Ed. 58(2019) 9496-9500. |
| [32] |
(a) Y. Xia, D. Qiu, J.B. Wang, Chem. Rev. 117(2017) 13810-13889; (b) M.P. Doyle, R. Duffy, M. Ratnikov, L. Zhou, Chem. Rev. 110(2010) 704-724; (c) Q.Q. Cheng, M.P. Doyle, Adv. Organomet. Chem. 66(2016) 1-31; (d) Z.F. Liu, X.F. Yue, Q. Wei, K.L. Han, Chin. Chem. Lett. 18(2007) 107-110. |
| [33] |
(a) Z. Sheng, Z.K. Zhang, C.H. Chu, et al., Tetrahedron 73(2017) 4011-4022; (b) X.Y. Wang, X. Wang, J.B. Wang, Tetrahedron 75(2019) 949-964; (c) Z.K. Zhang, Z. Sheng, W.Z. Yu, et al., Nat. Chem. 9(2017) 970-976. |
| [34] |
(a) X.H. Liu, H.F. Zheng, Y. Xia, et al., Acc. Chem. Res. 50(2017) 2621-2631; (b) X.B. Lin, Y. Tang, W. Yang, et al., J. Am. Chem. Soc. 140(2018) 3299-3305; (c) X. Lin, W. Yang, W.K. Yang, et al., Angew. Chem. Int. Ed. 58(2019) 13492-13498. |
| [35] |
L.K. Meng, P. Wu, J. Fang, et al., J. Am. Chem. Soc. 141 (2019) 11775-11780. DOI:10.1021/jacs.9b04619 |
| [36] |
(a) X.F. Xu, C. Li, Z.H. Tao, Y.J. Pan, Green Chem. 19(2017) 1245-1249; (b) X.F. Xu, C. Li, M.T. Xiong, et al., Chem. Commun. (Camb. ) 53(2017) 6219-6222; (c) X.J. Yan, C. Li, X.F. Xu, et al., Tetrahedron 75(2019) 3081-3087. |
| [37] |
(a) R. Hommelsheim, Y.J. Guo, Z. Yang, et al., Angew. Chem. Int. Ed. 58(2019) 1203-1207; (b) S. Jana, Z. Yang, C. Pei, et al., Chem. Sci. 10(2019) 10129-10134; (c) Z. Yang, Y.J. Guo, R.M. Koenigs, Chem. Eur. J. 25(2019) 6703-6706; (d) S. Jana, R.M. Koenigs, Asian J. Org. Chem. 8(2019) 683-686; (e) C. Empel, R.M. Koenigs, J. Flow Chem. 10(2020) 157-160. |
| [38] |
J.H. Yang, J.Z. Wang, H.T. Huang, et al., Org. Lett. 21 (2019) 2654-2657. DOI:10.1021/acs.orglett.9b00647 |
| [39] |
K. Orłowska, K. Rybicka-Jasinska, P. Krajewski, D. Gryko, Org. Lett. 22 (2020) 1018-1021. DOI:10.1021/acs.orglett.9b04560 |
| [40] |
(a) J.R. Shi, Y.Y. Li, Y. Li, Chem. Soc. Rev. 46(2017) 1707-1719; (b) T. Roy, A.T. Biju, Chem. Commun. (Camb. ) 54(2018) 2580-2594; (c) T. Matsuzawa, S. Yoshida, T. Hosoya, Tetrahedron Lett. 59(2018) 4197-4208; (d) Y.W. Zeng, J.B. Hu, Synthesis 48(2016) 2137-2150; (e) H. Yoshida, K. Takaki, Synlett 23(2012) 1725-1732; (f) D.B. Werz, A.T. Biju, Angew. Chem. Int. Ed. 59(2020) 3385-3398. |
| [41] |
(a) J.H. Chen, V. Palani, T.R. Hoye, J. Am. Chem. Soc. 138(2016) 4318-4321; (b) X. Xiao, T.R. Hoye, Nat. Chem. Biol. 10(2018) 838-844; (c) S.P. Ross, T.R. Hoye, Nat. Chem. Biol. 9(2017) 523-530. |
| [42] |
Y.M. Li, C. Mück-Lichtenfeld, A. Studer, Angew. Chem. Int. Ed. 55 (2016) 14435-14438. DOI:10.1002/anie.201608144 |
| [43] |
(a) X.B. Xu, Z.H. Lin, Y.Y. Liu, et al., Org. Biomol. Chem. 15(2017) 2716-2720; (b) M. Thangaraj, R.N. Gaykar, T. Roy, A.T. Biju, J. Org. Chem. 82(2017) 4470-4476; (c) J.J. Tan, T.Y. Zheng, K. Xu, C.Y. Liu, Org. Biomol. Chem. 15(2017) 4946-4950. |
| [44] |
T.Y. Zheng, J.J. Tan, R. Fan, et al., Chem. Commun. (Camb.) 54 (2018) 1303-1306. DOI:10.1039/C7CC08553B |
| [45] |
(a) R. Fan, B.B. Liu, T.Y. Zheng, et al., Chem. Commun. (Camb. ) 54(2018) 7081-7084; (b) H. Jian, Q. Wang, W.H. Wang, et al., Tetrahedron 74(2018) 2876-2883. |
| [46] |
W.H. Ding, A.M. Yu, L. Zhang, X.T. Meng, Org. Lett. 21 (2019) 9014-9018. DOI:10.1021/acs.orglett.9b03417 |
| [47] |
(a) F.L. Liu, J.R. Chen, Y.Q. Zou, Q. Wei, W.J. Xiao, Org. Lett. 16(2014) 3768-3771; (b) H.Y. Li, L.J. Xing, M.M. Lou, et al., Org. Lett. 17(2015) 1098-1101; (c) H. Hazatika, K. Neog, A. Sharma, et al., J. Org. Chem. 84(2019) 5846-5854; (d) Y.M. Li, A. Studer, Org. Lett. 19(2017) 666-669; (e) Y.Y. Li, D.C. Qiu, R.R. Gu, et al., J. Am. Chem. Soc. 138(2016) 10814-10817; (f) X.J. Li, Y. Sun, X. Huang, et al., Org. Lett. 19(2017) 838-841; (g) T. Matsuzawa, K. Uchida, S. Yoshida, T. Hosoya, Org. Lett. 19(2017) 5521-5524; (h) D.L. Chen, Y. Sun, M.Y. Chen, et al., Org. Lett. 21(2019) 3986-3989. |
| [48] |
(a) X.H. Ye, J. Wang, S.T. Ding, et al., Chem. Eur. J. 23(2017) 10506-10510; (b) J. Wang, S.Y. Zhang, C. Xu, et al., Angew. Chem. Int. Ed. 57(2018) 6915-6920. |
| [49] |
A. Parodi, S. Battaglioli, Y. Liu, et al., Chem. Commun. (Camb.) 55 (2019) 9669-9672. DOI:10.1039/C9CC04302K |
| [50] |
Z.Q. He, F.F. Song, H. Sun, Y. Huang, J. Am. Chem. Soc. 140 (2018) 2693-2699. DOI:10.1021/jacs.8b00380 |
| [51] |
M.T. Taylor, J.E. Nelson, M.G. Suero, M.J. Gaunt, Nature 562 (2018) 563-568. DOI:10.1038/s41586-018-0608-y |
| [52] |
(a) D.Y. Qian, J.W. Sun, Chem. Eur. J. 25(2019) 3740-3751; (b) V. Iaroshenko, Organophosphorus Chemistry: From Molecules to Applications, Wiley-VCH, Weinheim, 2019; (c) A. Golandaj, A. Ahmad, D. Ramjugernath, Adv. Synth. Catal. 359(2017) 3676-3706; (d) K. Ishihara, Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 85(2009) 290-313; (e) C.Q. He, C.C. Lam, P.Y. Yu, et al., J. Org. Chem. 85(2020) 2618-2625; (f) T. Nakamura, K. Okuno, R. Nishiyori, S. Shirakawa, Chem. Asian J. 15(2020) 463-472. |
| [53] |
S. kaneko, Y. Kumatabara, S. Shimizu, et al., Chem. Commun. (Camb.) 53 (2017) 119-122. DOI:10.1039/C6CC08411G |
| [54] |
(a) J.J. Tan, N. Yasuda, Org. Process Res. Dev. 19(2015) 1731-1746; (b) K. Maruoka, Proc. Jpn. Acad., Ser. B: Phys. Biol. Sci. 95(2019) 1-16; (c) R.J. Fox, J. Qiu, Org. Process Res. Dev. 24(2020) 235-241. |
| [55] |
S.Y. Liu, K. Maruoka, S. Shirakawa, Angew. Chem. Int. Ed. 56 (2017) 4819-4823. DOI:10.1002/anie.201612328 |
| [56] |
(a) J. Yoshida, A. Shimizu, Y. Ashikari, et al., Bull. Chem. Soc. Jpn. 88(2015) 763-775; (b) S. Suga, K. Matsumoto, K. Ueoka, J. Yoshida, J. Am. Chem. Soc. 128(2006) 7710-7711; (d) Y. Ashikari, T. Nokami, J. Yoshida, J. Am. Chem. Soc. 133(2011) 11840-11843; (e) R. Hayashi, A. Shimizu, J. Yoshida, J. Am. Chem. Soc. 138(2016) 8400-8403; (f) R. Hayashi, A. Shimizu, J.A. Davies, et al., Angew. Chem. Int. Ed. 57(2018) 12891-12895. |