 2021, Vol. 32
2021, Vol. 32 


b Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, School of Science, Tianjin University & Collaborative Innovation Center of Chemical Science and Engineering, Tianjin 300072, China;
c Hanshan Normal University, Chaozhou 521041, China;
d Key Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Key Laboratory of Molecule-Based Materials, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China;
e Department of Chemistry, Division of Science & Technology, University of Education, Lahore, Pakistan
Catalytic dehydrogenation of organic molecules is a fundamental and important process in the field of organic chemistry, which has attracted considerable attention in the field of H2 economy energy [1-3]. Homogeneous and heterogeneous catalysts could produce dehydrogenation reaction on some substrates, such as 1, 2, 3, 4-tetrahydroquinoline (THQ) and its derivatives [4, 5]. The dehydrogenation of alkanes includes two reaction paths: direct dehydrogenation and oxidative dehydrogenation [6, 7]. Direct dehydrogenation aims to produce H2 gas [8, 9]. Recently, using metal-based heterogeneous catalysts (including Cr, Pt, Pd3Au1, etc.) could dehydrogenate THQ [10-12]. Cr-based catalysts have low price and strong anti-poisoning ability, but have high biological toxicity. Pt-based catalysts have a unique ability to activate C—H bonds of alkanes with high activity but also have high price. Pt is easy to agglomerate in the process of catalyst cycle regeneration, which may cause irreversible deactivation of the catalyst. Thus, it is very valuable to expand the study of precious metal catalysts to non-precious metal catalysts, such as iron-base catalysts. For example, hierarchical dehydrogenation reactions could occur on a copper surface [13]. Cu-MnOX could be highly efficient oxidative dehydrogenation aromatization of THQ. [14] Single-atom site Cu could boost transferring hydrogenation of quinoline [15]. Co-N4 works as an efficient catalyst for dehydrogenation and transfer hydrogenation of N-heterocycles [16]. FeOX encapsulated in N-doped graphitic carbon provided a great performance towards dehydrogenation of N-heterocycles compounds [17, 18]. Iron-based nanocatalyst took place the acceptorless dehydrogenation reactions of N-heterocycles, alcohols and amines [19]. However, the mechanism of dehydrogenation on iron nanoparticles is not clear, which deserves to be further studied.
In this work, Fe NPs are adopted as an example to dehydrogenate mechanism of THQ which act as a probe molecule catalyst based on DFT. Fe NPs catalyst shows excellent catalytic performance for dehydrogenation of THQ with 100% selectivity and 100% conversion for 10—12 h at room temperature. Absorption energy of THQ and charge density difference are calculated to investigate the adsorption behavior and electronic properties. Finally, a clear energy profile with transition states is displayed to illustrate the dehydrogenation mechanism.
Synthesis of iron nanoparticles is as follows: In a typical synthesis, 12 g of melamine, 2 g of L-alanine, and 100 mg of Fe acetate were homogeneously mixed by ball milling for 1 h. Then, 15 mL of ethanol mixed with 3 mL of hydrochloric acid was added and the slurry was put in a mortar. The mixture was milled in a fume hood until all ethanol was evaporated. The resultant solid was dried in an oven at 60 ℃ overnight and ball milled again for 1 h. The thus obtained powder was pyrolyzed under flowing N2 atmosphere in a tube furnace with the following ramping program: from room temperature to 600 ℃ at a ramping rate of 2.5 ℃/min, then hold at 600 ℃ for 120 min, ramp to 900 ℃ at 5 ℃/min and hold for 90 min, finally the furnace was naturally cooled down to room temperature. The obtained black solid materials were grinded and then washed by 2 mol/L HCl aqueous solution at 80 ℃ for 24 h under stirring to remove metal particles. For copper-based material, 1 mol/L HNO3 was used to remove copper metal particles. The acid-washed material was dried and then annealed again in N2 at 800 ℃ for 1 h at a heating rate of 10 ℃/min to recover the crystallinity. The obtained black materials were marked as Fe NPs. The photos of Fe precursor, Fe NPs and washed liquid shown in Fig. S1 (Supporting information).
Catalytic dehydrogenation of THQ procedure is as follows: In a 5 mL glass vial, the magnetic stirring bar, the catalyst Fe nanoparticle (10 mg, 0.67 mol% Fe), THQ substrate (0.5 mmol) and solvent (3 mL) were added. The reaction was stirred at room temperature for 10 h. After completion of the reaction, the autoclave was cooled to room temperature, dodecane was added to the mixture as internal standard and the liquid was analyzed by gas chromatography (GC) to determine the conversion and selectivity.
Calculations were performed using spin-polarized Kohn-Sham density functional theory (DFT). We used the generalized gradient approximation with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation function as implemented in the Vienna ab initio simulation package (VASP) [20, 21]. The valence orbitals of Fe (3p, 3d), N (2s, 2p), C (2s, 2p), and H (1s) were described by plane-wave basis sets with cut-off energy of 400 eV. The Gaussian smearing method with a width of 0.20 eV was used. Spin was set as 2 and it is adjusted to consider the magnetism property of the Fe atoms. DFT + U (U = 3) overcomes the failure of DFT to describe Fe atoms whose 3d orbital is strong-correlated [22-26]. The Fe NPs were modeled with the vacuum gap set as 20 Å to avoid the interaction between the periodic images. The Brillouin zone was sampled at (2 × 2 × 1) for the calculations of Fe NPs surface, respectively. The convergence criteria for the energy and force were set to 10-4 eV and 0.02 eV/Å, respectively. In evaluating the energy barriers, all transition states and pathways were computed using the climbing image nudged elastic band (CI-NEB) method. The adsorption energies were calculated according to the equation, Eads = E(adsorbate/substrate) - [E(substrate) + E(adsorbate)], where E(adsorbate/substrate), E(adsorbate) and E(substrate) are energies of the substrate with the adsorbate, the gas-phase molecule and the clean substrate, respectively. The reaction energy and barrier were calculated by Er = E(FS) - E(IS) and Ea = E(TS) - E(IS), where E(IS), E(FS) and E(TS) are the energies of the corresponding initial state (IS), final state (FS), and transition state (TS), respectively.
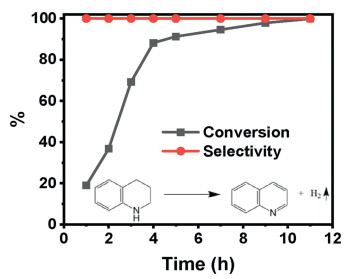
First, Fe NPs catalyst was tested for their reactivity toward dehydrogenation of THQ with the concomitant generation of dihydrogen under acceptor-free conditions at room temperature. The formula of THQ dehydrogenation is shown in insert picture of Fig. 1. Fe NPs catalyst has excellent dehydrogenation THQ performance with 100% selectivity and 100% conversion for 10-12 h at room temperature as shown in Fig. 1.

|
Download:
|
| Fig. 1. Conversion and selectivity of THQ catalysis on Fe NPs with an insert picture of THQ dehydrogenation formular. | |
{kind=link}
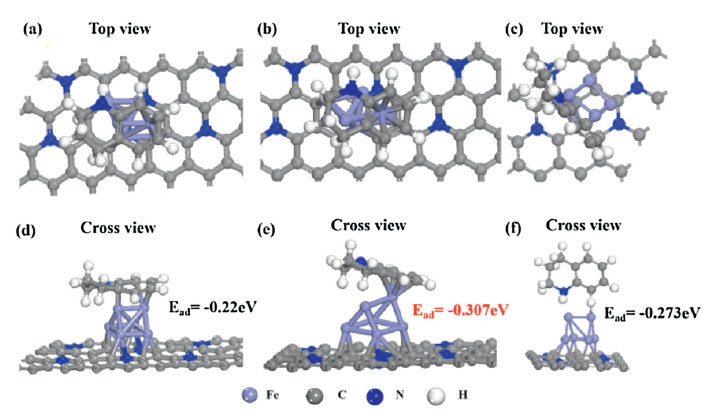
Then, theoretical calculations were conducted to shed light on the catalytic properties from electronic aspect on Fe NPs for acceptor dehydrogenation reaction. THQ was chosen as model absorbance, and the stable structures of Fe NPs, and corresponding THQ adsorption configuration and energy shown in Fig. 2 were all considered. In order to study the adsorption behavior of THQ on Fe NPs, two adsorption modes of THQ were taken into consideration, in which THQ molecular plane was parallel or perpendicular to the N-C surface. For the catalyst, all THQ prefers to be adsorbed with the molecular plane parallel to rather than perpendicular to the N-C surface. For example, the adsorption energy of THQ on Fe NPs N-C is -0.307 eV and -0.273 eV in parallel and perpendicular behavior, respectively (Fig. 2). Compared with Fe NPs N-C and catalyst without Fe (-0.02 eV, Fig. S2 in Supporting information), THQ adsorption energy on Fe NPs is smaller. A bar graph of the parallel or perpendicular adsorption energy comparison of THQ on N-C without Fe NPs and with Fe NPs is shown in Fig. S3 (Supporting information). Thus, the presence of Fe NPs shows great improvement for the adsorption ability for THQ, rationalizing the THQ dehydrogenation result.

|
Download:
|
| Fig. 2. The adsorption configurations and adsorption energy of THQ on Fe NPs-N-C in parallel and perpendicular behavior with (a–c) top view and (d–f) cross view. | |
{kind=link}
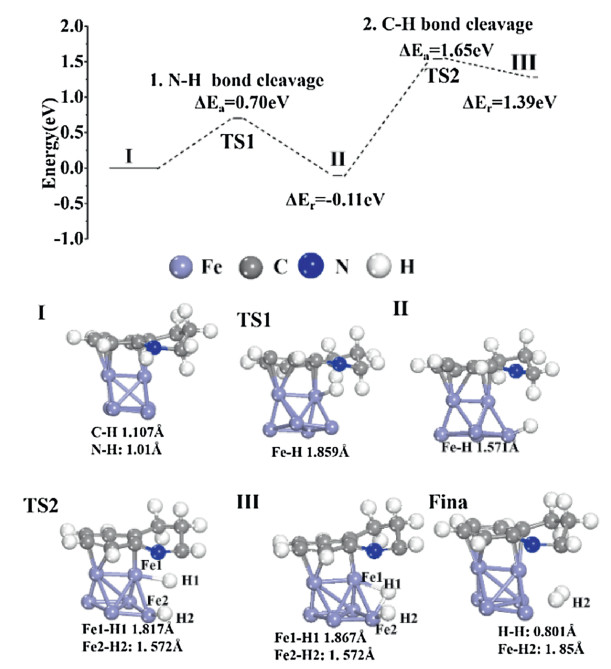
Importantly, a schematic figure of THQ dehydrogenation by Fe NPs is shown in Fig. S4 (Supporting information). Black Fe NPs powder is used to dehydrogenate THQ at the room temperature. A schematic diagram of THQ dehydrogenation is shown in Fig. S5 (Supporting information). First of all, the H connected to N is removed. Nitrogen and carbon form a double bond. Then the H of C—C on the ring where N is located is transferred to N—C, and the single bond becomes a double bond. Finally, N-C dehydrogenate the last H2. The energy profile with transition state of dehydrogenation catalytic reaction mechanism of THQ on the surface of Fe NPs is shown in Fig. 3. THQ is adsorbed horizontally on the surface of Fe NPs with adsorption energy being -0.307 eV. Firstly, N-H of THQ cleave with the energy barrier of 0.7 eV and it is an exothermic reaction. The bond length of Fe-H in the transition state TS1 is 1.859 Å. Then, the C—H bond in THQ breaks with the energy barrier of 1.65 eV and it is an endothermic reaction. The energy barrier is larger due to the large distortion of geometrical structure. The bond length of Fe1-H1 in transition state TS2 is 1.817 Å, and that of Fe2-N2 is 1.572 Å. Then, two hydrogen atoms are generated and escaped. A table of the bond length of construction of initial state, transition states and final states in energy profile of a catalytic reaction is shown in Table S1 (Supporting information). The distortion and the metastable adsorption would significantly increase the adsorption energy of the intermediate state and the H2 desorption behavior, which is the most important reason why Fe NPs show high catalytic activity.

|
Download:
|
| Fig. 3. The energy pathway of THQ catalysis on Fe NPs with corresponding detail configurations. | |
{kind=link}
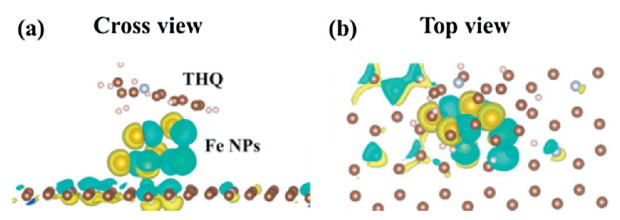
Charge density difference is also calculated to investigate the electrons transfer. For C-N without Fe catalyst, the THQ and carbon base would produce Π and Π adsorption as shown in Fig. S6 (Supporting information). Fe NPs could lose more electrons to carbon base as shown in Figs. 4a and b. An electron paramagnetic resonance (EPR) is conducted to characterize the ferromagnetic behavior of Fe NPs shown in Fig. S7 (Supporting information). The calculated g value equals to 2.0. The results show that the valence state of iron particles is Ⅲ valence, not zero valence.

|
Download:
|
| Fig. 4. Charge density difference (Δρ = ρ(adsorbate/substrate) - ρ(substrate) - ρ(adsorbate)) of THQ adsorption on Fe NPs with (a) cross view and (b) top view. The yellow region represents charge accumulation, and the cyan region indicates charge depletion. The brown color balls and light blue color balls represent C and N atoms. | |
{kind=link}
In conclusion, excellent performance in the dehydrogenation of THQ is demonstrated by Fe NPs catalyst with 100% selectivity and 100% conversion for 10-12 h at room temperature. A systemic mechanism study of THQ dehydrogenation is carried out on Fe NPs based on DFT. The calculated adsorption energy implies that THQ prefers to adsorb on Fe NPs. What is more, the energy barrier of transition state is relatively low, illustrating the dehydrogenation is feasible. This work provides an atomic scale mechanism guidance for the catalytic dehydrogenation reaction and points out the direction for the design of new catalysts.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21866032, 21805191) China Postdoctoral Science Foundation (No. 2020M672811) and the Guangdong Basic and Applied Basic Research Foundation (No. 2020A151501982). Our work is completed on the "Explorer 100" cluster system of Tsinghua National Laboratory for Information Science and Technology.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.10.040.
| [1] |
Y. Men, J. Su, X. Wang, et al., Chin. Chem. Lett. 30 (2019) 634-637. DOI:10.1016/j.cclet.2018.11.010 |
| [2] |
Z. Wang, E. Li, Z. He, J. Chen, Y. Huang, Acta Phys. Chim. Sin. 35 (2019) 906-912. DOI:10.3866/PKU.WHXB201811038 |
| [3] |
Z. Zhang, B. Xu, X. Wang, Chem. Soc. Rev. 43 (2014) 7870-7886. DOI:10.1039/C3CS60389J |
| [4] |
Y. Wu, Z. Chen, W.C. Cheong, et al., Chem. Sci. 10 (2019) 5345-5352. DOI:10.1039/C9SC00475K |
| [5] |
I. Muthukrishnan, V. Sridharan, J.C. Menendez, Chem. Rev. 119 (2019) 5057-5191. DOI:10.1021/acs.chemrev.8b00567 |
| [6] |
B. Yan, J. Sheng, B. Qiu, D. Wang, A. Lu, Sci. Sin. Chim. 50 (2020) 832. DOI:10.1360/SSC-2020-0014 |
| [7] |
F. Chen, S. Zhao, T. Yang, et al., Acta Phys. Chim. Sin. 35 (2019) 1422-1423. DOI:10.3866/PKU.WHXB201907026 |
| [8] |
T. Mitsudome, Y. Mikami, H. Funai, et al., Angew. Chem. Int. Ed. 47 (2008) 138-141. DOI:10.1002/anie.200703161 |
| [9] |
Z. Li, F. Zhai, H. Qiu, et al., Rare Met. 39 (2020) 383-391. DOI:10.1007/s12598-016-0711-x |
| [10] |
Y. Yu, X. Wang, C. Liu, et al., J. Energy Chem. 40 (2020) 212. DOI:10.1016/j.jechem.2019.04.017 |
| [11] |
A.M.S. Pembere, C. Cui, H. Wu, Z. Luo, Chin. Chem. Lett. 30 (2019) 1000-1004. DOI:10.1016/j.cclet.2018.12.019 |
| [12] |
F. Nosheen, N. Wasfi, S. Aslam, et al., Nanoscale 12 (2020) 4219-4237. DOI:10.1039/C9NR09557H |
| [13] |
K. Wu, Acta Phys. Chim. Sin. 35 (2019) 349-350. DOI:10.3866/PKU.WHXB201805023 |
| [14] |
M. Liu, Acta Phys. Chim. Sin. 35 (2019) 1041-1042. DOI:10.3866/PKU.WHXB201905045 |
| [15] |
J. Zhang, C. Zheng, M. Zhang, et al., Nano Res. 13 (2020) 3082-3087. DOI:10.1007/s12274-020-2977-4 |
| [16] |
Y. Han, Z. Wang, R. Xu, et al., Angew. Chem. Int. Ed. 57 (2018) 11262-11266. DOI:10.1002/anie.201805467 |
| [17] |
H. Wei, X. Liu, A. Wang, et al., Nat. Commun. 5 (2014) 5634. DOI:10.1038/ncomms6634 |
| [18] |
Z. Chen, W. Yang, Y. Wu, et al., Nano Res. 13 (2020) 3075-3081. DOI:10.1007/s12274-020-2975-6 |
| [19] |
G. Jaiswal, V.G. Landge, D. Jagadeesan, E. Balaraman, Nat. Commun. 8 (2017) 2147. DOI:10.1038/s41467-017-01603-3 |
| [20] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [21] |
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169-11186. DOI:10.1103/PhysRevB.54.11169 |
| [22] |
B. Qiao, A. Wang, X. Yang, et al., Nat. Chem. 3 (2011) 634-641. DOI:10.1038/nchem.1095 |
| [23] |
G. Rollmann, Phys. Rev. B 69 (2004) 165107. DOI:10.1103/PhysRevB.69.165107 |
| [24] |
J.J. Tang, B. Liu, J. Phys. Chem. C 120 (2016) 6642-6650. DOI:10.1021/acs.jpcc.6b00374 |
| [25] |
Z. Chen, Y. Mao, J. Chen, et al., ACS Catal. 7 (2017) 4281-4290. DOI:10.1021/acscatal.7b00541 |
| [26] |
G. Chen, Y. Zhao, G. Fu, et al., Science 344 (2014) 495-499. DOI:10.1126/science.1252553 |