 2021, Vol. 32
2021, Vol. 32 


b School of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, China;
c Instituto de Productos Naturales y Agrobiologi'a, Consejo Superior de Investigaciones Científicas (IPNA-CSIC), La Laguna 38206, Spain;
d Jiangsu Key Laboratory of Drug Screening, State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, Nanjing, 210009 China
Soft corals of the genus Sarcophyton (family Alcyoniidae), chemically and biologically investigated over decades, were regarded as rich sources of structurally diverse and biologically active terpenoids, e.g., cembranoids [1]. Although cembranoids were frequently encountered in marine Cnidaria, dimeric cembranoids (biscembranoids) were rare, with only approximate 70 examples been found in Nature, mostly in Sarcophyton soft corals [1-6]. Depending on the dimerization patterns of the Diels-Alder reaction over monomeric cembranoids, biscembranoids mainly comprise three types, including ximaolides [7, 8], bisglaucumlides [9] and bislatumlides [10]. A wide range of biological activities, such as cytotoxic, antimicrobial and anti-inflammatory effects, have been reported for these biscembranoids [11]. South China Sea breeds abundant marine life, including over 40 identified species of Sarcophyton soft corals, of which nearly 20 species were chemically investigated and about half of them were reported by our group. In fact, our group has long been engaged in the chemical and biological studies on marine invertebrates from South China Sea, and numerous complex and bioactive terpenoids were isolated and structurally determined [12, 13, 14a, b, 15-17]. In particular, over one third of the natural biscembranoids have been discovered by our group, such as ximaolides A–G [7, 8], and bislatumlides C–F [10], which were isolated from S. tortusum and S. latum of previous collections.
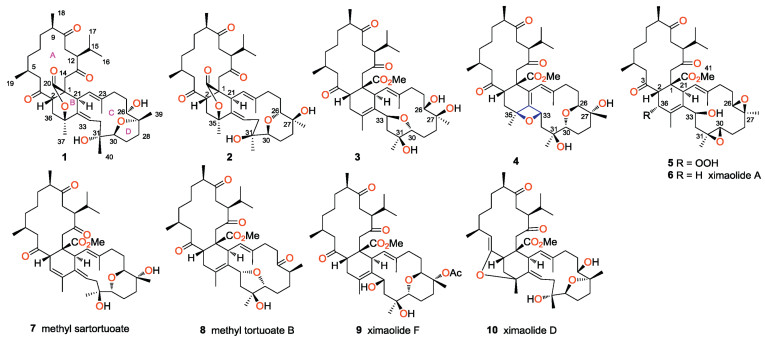
In order to expand the structural diversity and pharmacological applications of biscembranoids from S. tortusum, as well as to deal with the challenging configurational problems of several complex molecules, the title animals were recollected from Yalong Bay, Hainan, China. A systematic chemical investigation has led to the isolation and structural determination of five novel biscembranoids, ximaolides H-L (1-5), together with four known related ones (6-9) (Fig. 1). Herein, we report the isolation, structural elucidation, and biological activity evaluation of the isolated metabolites.

|
Download:
|
| Fig. 1. Structures of compounds 1 - 10. | |
The frozen title animals (dry weight 100 g) were cut into pieces and exhaustively extracted with acetone. The Et2O-soluble portion of the acetone extract was repeatedly column chromatographed over silica gel, Sephadex LH-20, and RP-HPLC to yield nine pure compounds 1-9, respectively. The structures of the known compounds were readily identified as ximaolide A (6) [7], methyl sartortuoate (7) [18], methyl tortuoate B (8) [19] and ximaolide F (9) [8], respectively, by direct comparison of their NMR data and CD spectra with those reported in the literature.
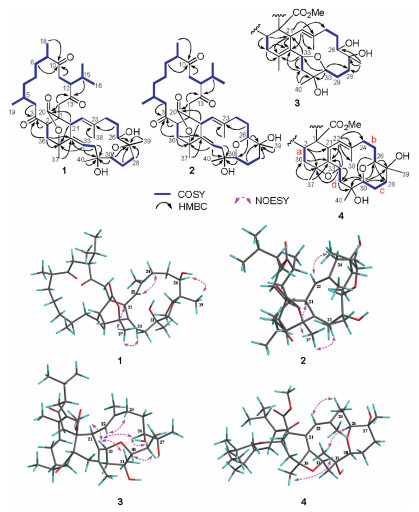
Ximaolide H (1) was obtained as a white amorphous powder. Its molecular formula was deduced to be C40H60O8 by HREI-MS from the molecular ion peak at m/z 668.4295 [M]+ (calcd. 668.4288), indicating eleven degrees of unsaturation. The analysis of 13C NMR data, DEPT and HSQC spectra of 1 revealed 40 carbon signals, including three ketone carbonyl groups (δC 214.5, 213.6, 209.3), an ester carbonyl group (δC 173.6), four olefinic carbons (δC 139.4, 138.8, 121.9, 121.7), five oxygenated carbons (δC 87.1, 83.9, 82.5, 73.8, 71.9), and eight methyls (δC 22.5, 22.3, 22.2, 21.0, 20.3, 19.4, 17.4, 17.3) (Table 1). Four carbonyls and two double bonds accounted for six degrees of unsaturation, indicating that 1 contained a pentacyclic system. These NMR data were reminiscent of those of the co-isolated known compounds 6-9, suggesting that ximaolide H (1) possesses a biscembranoid skeleton. In particular, the similar NMR signals from C-3 to C-19 between 1 and 6-9 indicated that they should share a common ring A. Further literature survey and careful data comparison indicated that 1 should comprise the same rings C and D as ximaolide D (10) [7], a biscembranoid previously isolated by our group from the Hainan soft coral S. tortuosum, since they showed very similar NMR signals from C-21 to C-34. Therefore, the remaining task to identify the planar structure of compound 1 was the substitution patterns on ring B. The only significant differences between 1 and ximaolide D on ring B were the lack of a carbomethoxy group at C-1 in 1 and the downfield shift of C-35 from δC 76.9 in ximaolide D to 82.5 in 1, suggesting that the methyl ester at the C-1 position of ximaolide D was replaced by an 135-bridged lactone moiety on ring B of 1. The proposed planar structure of 1 was further confirmed by detailed analysis of its 2D NMR spectra, including 1H-1H COSY and HMBC experiments, as shown in Fig. 2. In particular, the key HMBC correlations from H2-14 (δH 3.20, 3.02) and H-21 (δH 4.30) to C-20 (δC 173.6); from H-21, H-33 (δH 5.35), and H2-36 to C-35 (δC 82.5), supported the linkage position of the 135-bridged lactone.
|
|
Table 1 The 13C NMR data of compounds 1-5 in CDCl3a. |
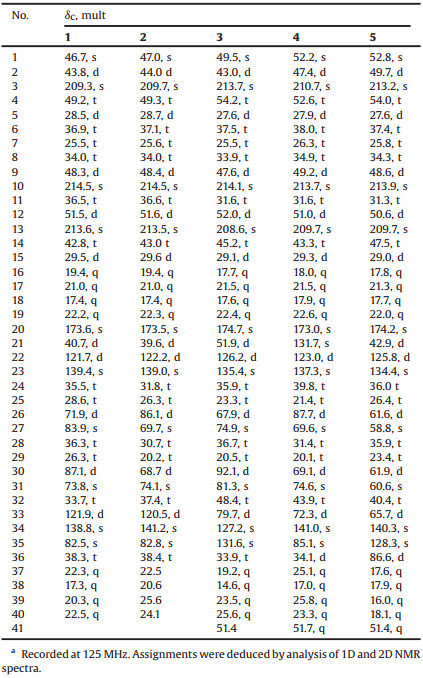

|
Download:
|
| Fig. 2. 1H-1H COSY, key HMBC and NOESY correlations of compounds 1 - 4. | |
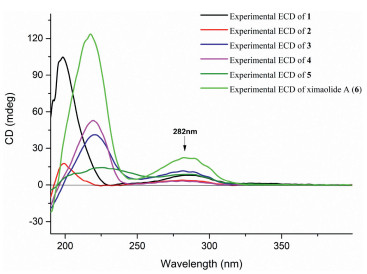
The E geometry of Δ3334 was determined by the clear NOE correlation between H-33 and H3-37 (δH 1.52), whereas the absence of the NOE cross peak between H-22 (δH 4.55) and H3-38 (δH 1.91) and the clear NOE relationship between H-22 and H2-24 (δH 2.17) (Fig. 2) indicated the E geometry of Δ22,23. According to our previous research, the three isolated carbonyl groups in ring A of ximaolides A - C and E - G [7, 8, 20], due to the n-π* transitions, resulted in a positive Cotton effect at 282 nm in its CD spectrum (Fig. 3), which further influenced the stereochemistry of the adjacent chiral carbons C-1, C-2, C-5, C-9, C-12 and C-21.

|
Download:
|
| Fig. 3. Experimental ECD spectra of 1 - 6. | |
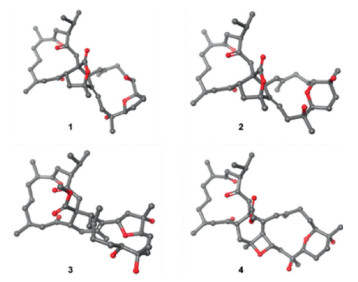
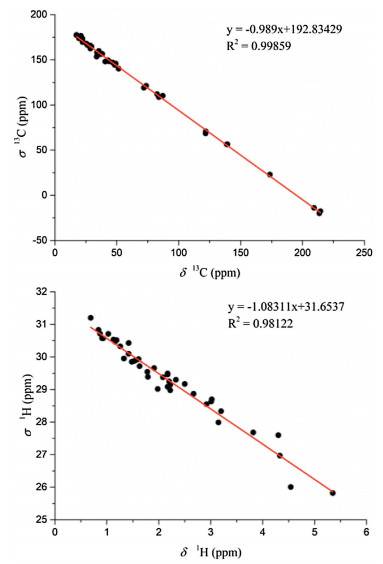
Therefore, the ACs of C-1, C-2, C-5, C-9, C-12, and C-21 in ximaolide H (1) were determined by showing the same positive cotton effect at 282 nm in its CD spectrum as those of ximaolides A, D and F, to be 1S, 2S, 5S, 9R, 12S, 21S (Fig. 3). The configuration of chiral center C-35 was assigned to be S by the obvious NOE relationship between H-21 and H3-37. The major difficulty was to connect the relative configuration of the chiral centers of rings C/D with the remote position C-35. This problem has been tackled by using J-based conformational analysis and NOE analysis. The scarcity of informative NOE correlations led us to find feasible alternatives for structural determination. Computational prediction of NMR data is clearly becoming a fast and reliable approach for structure elucidation of natural products [21]. Therefore, the relative configuration of 1 was further deduced by quantum mechanics calculations of its NMR data (QM-NMR). First, conformational searches on the 32 possible candidate diastereoisomers (Fig. S43 in Supporting information) at the Merck Molecular Force Field (MMFF) as implemented in the Macromodel software were performed. All structures found within an energy window of 21 kJ/mol were saved, using an RMSD = 1.0 A. Next, geometrical optimization followed by NMR calculations at the PCM/mPW1PW91/6-31+G**//B3LYP/6-31G* level of theory was done, the most stable conformation by DFT calculations of compound 1 were shown in Fig. 4. NMR shielding constants were calculated using the GIAO approach [22] and were averaged over the Boltzmann distribution estimated for each stereoisomer. Finally, the computed and experimental data were correlated and their corresponding DP4+ probabilities were estimated. As a result, it turned out that the experimental NMR data of compound 1 gave the best match for isomer 1S*, 2S*, 5S*, 9R*, 12S*, 21S*, 26R*, 27R*, 30S*, 31R*, 35S* with 99% probability. The selected stereoisomer showed linear correlation coefficients (R2) of 0.999 and 0.981 (Fig. 5) and CMAE (corrected mean absolute error) values of 1.8 and 0.12 ppm (Table S1 in Supporting information), for carbon and proton data, respectively. Thus, the AC of compound 1 was determined as 1S, 2S, 5S, 9R, 12S, 21S, 26R, 27R, 30S, 31R, 35S.

|
Download:
|
| Fig. 4. Most stable conformations by DFT calculations of compounds 1 - 4. | |

|
Download:
|
| Fig. 5. Correlation plots between 13C/1H isotropic magnetic shielding values (σ) computed at the PCM/mPW1PW91/6-31+G** level of theory and experimental 13C/1H chemical shifts (δ) for compound 1. | |
Compound 2, named ximaolide I, was also obtained as white powder. Its HRMS (ESI-TOF) m/z: [M + Na]+ calcd. for C40H60O8Na 691.4186; Found 691.4184, revealed the molecular formula of C40H60O8, which is the same as that of 1, suggesting that they are isomers. Its 1H and 13C NMR data (Table 1, Table 2) were also similar to those of compound 1, especially those in rings A and B, indicating the same biscembranoid skeleton with the same ring A and 135-bridged lactone in ring B. The major differences between these two compounds were found to be in the NMR signals from C-25 to C-30, suggesting that the tetrahydrofuran ring of 1 was replaced by a tetrahydropyran ring D in 2, the same as that of methyl sartortuoate (7) [18], with the clear 1H-1H COSY and HMBC correlations as shown in Fig. 2. The E geometry of Δ22,23 and Δ33,34, and the configurations of the chiral centers in rings A - C of 2 were deduced to be the same as those of 1 by NOESY experiments (Fig. 2) and by the same positive Cotton effects of their CD spectra (Fig. 3). The configurations of the chiral centers on rings C and D were also determined by the QM-NMR due to the lack of informative NOE relationships. Again, making use of a QM-NMR approach, 32 relative isomers (Fig. S44 in Supporting information) of 2 were built and conformational searches followed by DFT optimization (Fig. 4) and calculation of their NMR parameters were undertaken. The candidate structure with a relative configuration 1S*, 2S*, 5S*, 9R*, 12S*, 21S*, 26R*, 27S*, 30S*, 31S*, 35S* gave a best match between experimental and computed data with 99% probability (CMAE of 2.1 and 0.20 ppm).
|
|
Table 2 The 1H NMR data of compound 1 - 5 in CDCl3.a |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Based on the above evidence and on biogenetic considerations, the absolute stereochemistry of compound 2 was determined as 1S, 2S, 5S, 9R, 12S, 21S, 26S, 27R, 30R, 31S, 35S.
Ximaolide J (3) was obtained as white powder. Its HREI-MS at m/z 700.4555 [M]+ (calcd. for C41H64O9, 700.4550) revealed the molecular formula of C41H64O9, with ten degrees of unsaturation. The 1H and 13C NMR data (Table 1, Table 2) indicated the presence of a methyl ester [δC 174.7 (C), 51.4 (CH3) and δH 3.52 (3H, s)], three ketone carbonyls (δC 213.7, 214.1, 208.6), one tri-substituted double bond [δC/H 126.2/4.94 (CH), 135.4 (C)] and one tetra-substituted double bond [δC 127.2 (C), 131.6 (C)], five oxygenated carbons [δC/H 74.9 (C), 81.3 (C), 67.9/3.14 (CH), 79.7/3.93(CH), 92.1/3.99(CH)], and eight methyl groups (δC/H 17.7/0.70, 21.5/1.01, 17.6/1.12, 22.4/0.85, 19.2/1.89, 14.6/1.59, 23.5/1.14, 25.6/1.39). All the above data, together with the 2D NMR analysis (Fig. 2), suggested that compound 3 is a biscembranoid, with a structure similar to the co-isolated methyl tortuoate B (8) [19]. The main differences between these two compounds occurred on C-26 and C-27. In particular, the secondary hydroxyl at C-26 (δC/H 67.9/3.14) and the tertiary hydroxyl at C-27 (δC 74.9) of 3 replaced the carbonyl at C-26 (δC 215.9) and the methine at C-27 (δC/H 49.0/2.37) of 8. The stereochemistry on rings A and B was determined to be the same as the co-isolated 7-9 by the similar positive Cotton effects at 282 nm in their CD spectra. The E geometry of Δ22,23 was also deduced by the absence of the NOE cross peaks between H-22 (δH 4.94) and H3-38 (δH 1.59), and the clear NOE correlation of H-22 and H2-24 (δH 2.29, 2.12). Furthermore, the clear NOE correlations of H-21 (δH 2.61) and H-33 (δH 3.93), H-33 and H-26 (δH 3.14), H-33 and H3-40 (δH 1.39) determined the relative configuration at C-21, C-26, C-31, and C-33 as 21S*, 26S*, 31S*, 33S*, while the configuration at C-27 and C-30 was uncertain because of lacking of key NOE signals. Thus, the whole relative configuration of 3 was also elucidated via QM-NMR calculations, using the same protocol used for 1 and 2. 64 candidate diasteroisomers (Fig. S45 in Supporting information) were studied and this time the calculations gave a best match for isomer 1S*, 2S*, 5S*, 9R*, 12S*, 21S*, 26S*, 27S*, 30R*, 31S*, 33S* with more than 99% probability (Fig. 4). CMAE values of 1.9 and 0.16 ppm for carbon and proton data, respectively, were obtained.
Based on these evidences and on biogenetic considerations, the absolute configuration of compound 3 was assigned as 1S, 2S, 5S, 9R, 12S, 21S, 26S, 27S, 30R, 31S, 33S as shown in Fig. 1.
The molecular formular of ximaolide K (4) was deduced as C41H62O9 by HREI-MS at m/z 698.4401 [M]+, with eleven degrees of unsaturation. The 1H and 13C NMR spectra (Table 1, Table 2), as well as an HSQC experiment, indicated the presence of a methyl ester [δC 173.0 (C), 51.7 (CH3) and δH 3.48 (3H, s)], three ketone carbonyl groups (δC 210.7, 213.7, 209.7), one tri-substituted double bond [δC/H 123.0/5.63 (CH), 137.3 (C)] and one tetra-substituted double bond [δC 131.7 (C), 141.0 (C)], six oxygenated carbons [δC/H 87.7/3.58 (CH), 69.6 (C), 74.6 (C), 69.1/3.55 (CH), 72.3/4.77 (CH), 85.1 (C)], and eight methyl groups (δC/H 18.0/0.74, 21.5/0.98, 17.9/1.11, 22.6/0.87, 25.1/1.59, 17.0/1.56, 25.8/1.10, 23.3/1.18). All the above signals indicated that compound 4 is also a biscembranoid, with the same ring A as the other co-isolated compounds. Four carbonyls and two double bonds accounted for six degrees of unsaturation, thus the remaining five degrees should be ascribed to a pentacyclic ring system. The 1H-1H COSY correlations of H-2 (δH 3.86)/H2-36 (δH 2.57, 1.69), H2-24 (δH 2.32, 2.18)/H2-25 (δH 1.89, 1.47)/H-26 (δH 3.58), H2-28 (δH 1.93, 1.56)/H2-29 (δH 1.75)/H-30 (δH 3.55) and H2-32 (δH 2.03, 1.94)/H-33 (δH 4.77) disclosed the proton connectivity for four structural fragments a-d (Fig. 2). Fragment a, along with the HMBC correlations from H-2 to C-21 (δC 131.7)/C-35 (δC 85.1), H2-36 to C-34 (δC 141.0)/C-35 (δC 85.1), H3-37 (δH 1.59) to C-34/C-36 (δC 34.1), constituted the six membered ring B, with the tetra-substituted double bond located at Δ21,34. Fragments b-d, together with the HMBC correlations from H-22 (δH 5.63) to C-23 (δC 137.3)/C-24 (δC 39.8)/C-34/C-38 (δC 17.0), H-26 to C-27 (δC 69.6)/C-28 (δC 31.4), H-30 to C-28/C-31 (δC 74.6)/C-32 (δC 43.9), H2-32 to C-31, H-33 to C-21/C-32/C-34/C-35, constructed the 14-membered cembrane ring C, with the location of the trisubstituted double bond at Δ22,23. The biscembranoid skeleton of 4 was then set up, and the remaining two rings must be attributed to two cyclic ethers. The first cyclic ether was deduced to be a tetrahydropyran ring D, connecting C-26 and C-30, since the HMBC correlations from H-26 to C-30 and from H-30 to C-26 have been both observed (Fig. 2). The extremely similar 13C NMR data of C-26/C-27/C-30/C-31 (87.7/69.6/69.1/74.6) with those of the co-occurring compounds 7 and 9, not only confirmed the presence of the tetrahydropyran ring D, but also indicated that the two hydroxyls should be located at C-27 and C-31. Therefore, the last cyclic ether ring must be constructed by a connection of C-33 and C-35, forming an oxetane ring. Finally, the planar structure of compound 4 was determined as shown in Fig. 1. The relative configuration of 4 was assigned by NOESY correlations and by comparison with the co-isolated compounds 7 and 9 which contain the same tetrahydropyran ring. The relative configuration of chiral centers C-26, C-33 and C-35 were determined to be 26S*, 33S*, 35S* by the distinct NOE correlations of H-26 (δH 3.58) and H-33 (δH 4.77), H-33 and H3-37 (δH 1.59). The whole relative configuration of 4 was further elucidated via QM-NMR calculations, using the DP4+ protocol as used for 1–3. This time the calculations gave a best match for isomer 1S*, 2S*, 5S*, 9R*, 12S*, 26S*, 27R*, 30R*, 31S*, 33S*, 35S* with 80% probability (Figs. 4 and Fig. S50 in Supporting information). CMAE values of 2.9 and 0.17 ppm for carbon and proton data, respectively, were obtained.
Based on these evidences and on biogenetic considerations, the absolute configuration of compound 4 was deduced as 1S, 2S, 5S, 9R, 12S, 26S, 27R, 30R, 31S, 33S, 35S as shown in Fig. 1.
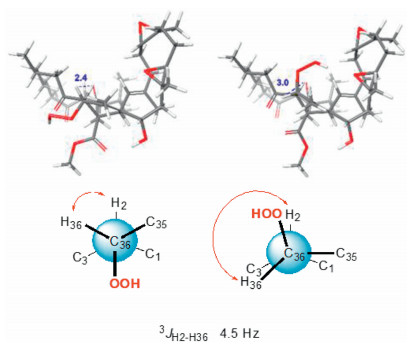
Ximaolide L (5) was obtained as colorless oil. The HREI-MS at m/z 714.4318 [M]+ (calcd. for C41H62O10, 714.4343) indicated the molecular formula as C41H62O10. Its 1H and 13C NMR data (Table 1, Table 2) were extremely similar to those of co-isolated ximaolide A (6). In fact, the only difference between 5 and 6 was the presence of a peroxide at the C-36 position of 5, since the 13C NMR signal of C-36 has been shifted from δC 33.0 (CH2) in 6 to δC 86.6 (CH) in 5 and there are two additional oxygens in the composition of 5. The obvious NOE correlations between the adjacent H-2 (δH 4.20) and H-36 (δH 4.68) on the cyclohexene ring B assigned the configuration of C-36. Moreover, thanks to 1H NMR spectroscopic analysis, it is possible to observe a coupling constant between H-2 and H-36 (3J2,36 = 4.5 Hz) which suits better with the isomer configuration 36R* (Fig. 6) as it is described in Karplus equation. Thus, the α-orientation of the peroxide group was determined. All the other configurations of 5 were determined to be the same as those of 6 by their similar NMR data around the chiral centers and similar CD spectra with the same positive Cotton effects at 282 nm.

|
Download:
|
| Fig. 6. The determination of the configuration at C-36 of 5. | |
{kind=link}
In the biological activity test, the new compounds 1-5 gave negative results in both antibacterial and cytotoxic effects, whereas in the anti-inflammatory assay, ximaolides I (2), K (4), and F (9) exhibited significant inhibition of LPS-induced TNF-α protein release in RAW264.7 macrophages at a concentration of 20 μmol/L (Fig. 7). In addition, none of the compounds showed obvious cytotoxicity against RAW264.7 cells, with inhibition ratio less than 50% at a concentration of 40 μmol/L.

|
Download:
|
| Fig. 7. Effects of the bioactive compounds 2, 4 and 9 on LPS-induced inflammatory response in RAW264.7 macrophages. Results are expressed as mean ± SEM. **P < 0.01, ***P < 0.001 vs. the LPS group μM: μmol/L. | |
{kind=link}
In summary, five new (1-5) and four known (6-9) biscembranoids were isolated and characterized from the Hainan soft coral S. tortuosum collected off Yalong Bay. Among them, compounds 1 and 2, containning 1,35-bridged lactone moiety in ring B, and compound 5, comprising a peroxide group, are unprecedented natural biscembranoids. The discovery of such novel structure skeleton has further expanded the chemical diversity and complexity of marine terpenoids, which was probably influenced by the different living environment of the title animals, since they were collected in a different water area as our previously reported ones [7, 8]. It is worth mentioning that the determination of configuration is a big challange for cembranoids and biscembranoids, since NOESY experiments are not 100% reliable due to the conformation flexibility of the macrocyle in these compounds. The stereochemistry of the reported new compounds 1-5 were determined by a combination of NOESY experiments, QM-NMR calculations, ECD spectra comaprison, and biogenetic considerations. In the bioassay, it is suprising that the new compounds did not show cytotoxicity or antimicrobial activity as did the previouly reported analogs, while anti-inflammatory activity of the new compounds were observed, which may give an insight for related drug discovery.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis research work was financially supported by the National Natural Science Foundation of China (NSFC, Nos. 81991521, 8202290170, 42076099), the NSFC/CNRS (Centre National de la Recherche Scientifique) Joint Project (No. 81811530284), the National Key Research and Development Program of China (No. 2018YFC0310903), and the SKLDR/SIMM Project (No. SIMM1903ZZ-04). X.-W. Li is also thankful for the Shanghai Rising-Star Program (No. 20QA1411100) and SA-SIBS Scholarship Program. We thank Prof. X.-B. Li from Hainan University for the taxonomic identification of the soft coral material.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.037.
| [1] |
L.F. Liang, Y.W. Guo, Chem. Biodivers. 10 (2013) 2161-2196. DOI:10.1002/cbdv.201200122 |
| [2] |
P. Sun, F.Y. Cai, G. Lauro, et al., J. Nat. Prod. 82 (2019) 1264-1273. DOI:10.1021/acs.jnatprod.8b01037 |
| [3] |
W. Li, Y.H. Zou, M.X. Ge, et al., Mar. Drugs 15 (2017) 85. DOI:10.3390/md15040085 |
| [4] |
P. Sun, Q. Yu, J. Li, et al., J. Nat. Prod. 79 (2016) 2552-2558. DOI:10.1021/acs.jnatprod.6b00453 |
| [5] |
C.Y. Huang, P.J. Sung, C. Uvarani, et al., Sci. Rep. 5 (2015) 15624. DOI:10.1038/srep15624 |
| [6] |
H.N. Nguyen, P.T. Tung, N.T. Ngoc, et al., Chem. Pharm. Bull. 63 (2015) 636-640. DOI:10.1248/cpb.c15-00273 |
| [7] |
R. Jia, Y.W. Guo, P. Chen, et al., J. Nat. Prod. 70 (2007) 1158-1166. DOI:10.1021/np060220b |
| [8] |
R. Jia, Y.W. Guo, E. Mollo, et al., Helv. Chim. Acta 91 (2008) 2069-2074. DOI:10.1002/hlca.200890220 |
| [9] |
T. Iwagawa, K. Hashimoto, H. Okamura, et al., J. Nat. Prod. 69 (2006) 1130-1133. DOI:10.1021/np058115+ |
| [10] |
R. Jia, T. Kurtán, A. Mándi, et al., J. Org. Chem. 78 (2013) 3113-3119. DOI:10.1021/jo400069n |
| [11] |
I.G. Rodrigues, M.G. Miguel, W. Mnif, Molecules 24 (2019) 781. DOI:10.3390/molecules24040781 |
| [12] |
M. Yang, X.L. Li, J.R. Wang, et al., J. Org. Chem. 84 (2019) 2568-2576. DOI:10.1021/acs.joc.8b03020 |
| [13] |
G. Li, H. Li, Q. Zhang, et al., J. Org. Chem. 84 (2019) 5091-5098. DOI:10.1021/acs.joc.9b00030 |
| [14] |
(a) F. Ye, J. Li, Y. Wu, et al., Org. Lett. 20 (2018) 2637-2640; (b) F. Ye, Z.D. Zhu, J.S. Chen, et al., Org. Lett. 19 (2017) 4183-4186. |
| [15] |
S.W. Li, F. Ye, Z.D. Zhu, et al., Acta Pharm. Sin. B 8 (2018) 944-955. DOI:10.1016/j.apsb.2018.06.004 |
| [16] |
Q.H. Wu, S.W. Li, H. Xu, et al., Angew. Chem. Int. Ed. 59 (2020) 12105-12112. DOI:10.1002/anie.202003643 |
| [17] |
Y.F. Li, L.L. He, H.L. Liu, et al., J. Asian Nat. Prod. Res. 15 (2013) 566-573. DOI:10.1080/10286020.2013.787988 |
| [18] |
P. Yan, Z. Deng, L. van Ofwegen, et al., Chem. Biodivers. 8 (2011) 1724-1734. DOI:10.1002/cbdv.201000244 |
| [19] |
L.M. Zeng, W.J. Lan, J.Y. Su, et al., J. Nat. Prod. 67 (2004) 1915-1918. DOI:10.1021/np0400151 |
| [20] |
T. Kurtán, R. Jia, Y. Li, et al., Eur. J. Org. Chem. 2012 (2012) 6722-6728. DOI:10.1002/ejoc.201200862 |
| [21] |
M.W. Lodewyk, M.R. Siebert, D.J. Tantillo, Chem. Rev. 112 (2012) 1839-1862. DOI:10.1021/cr200106v |
| [22] |
N. Grimblat, A.M. Sarotti, Chem. Eur. J. 22 (2016) 12246-12261. DOI:10.1002/chem.201601150 |