 2021, Vol. 32
2021, Vol. 32 


b Laboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China;
c Shandong Center for Food and Drug Evaluation and Certification, Ji'nan 250013, China
Nanotechnology is an upcoming and rapidly expanding field, which affects our lives tremendously over the next decade in different fields, especially in medicine. Nanomedicine provides significant improvements in the diagnosis, treatment and management of diseases, contributing to realize the personalized medicine [1-4]. By 2017, 51 products based on this technology have been applied in clinical practice, and 1500 patents have been completed [5-8]. Among the different nanotherapeutics, polymeric nanoparticles have made a significant impact in the clinic [9].
Nanotechnology was used in pharmaceutics to improve the delivery of poorly soluble, highly-toxic or unstable drugs [9-11]. PM are recognized as one of the most promising drug carriers in clinical translation with the high rate of regulatory authorities approval, which are the inner core/outer shell nanostructure with a diameter of several tens of nanometers (10-100 nm) assembled by amphiphilic block copolymers in the aqueous solution (Fig. 1) [12-16]. Active pharmaceutical ingredients (API) were incorporated into the inner hydrophobic core of PM, either by chemical conjugation or by physical entrapment [17-20]. Moreover, the outer hydrophilic shell of PM can be further functionalized with different moieties such as environment-responsive cleavable linkages or targetable ligands, which will further improve specificity and efficacy of micelle-based drug delivery [21-24]. The clinical application of PM formulations is mostly through intravenous administration and intended for the delivery of anticancer agents.

|
Download:
|
| Fig. 1. Self-assembled PM as intelligent nanocarriers for drug delivery. The incorporated API is released from the inner core by diffusion. | |
Several PM have already entered clinical practice, while most are currently in clinical investigation for multiple indications [25, 26]. Notably, PM can solve the main drawbacks of conventional medicines such as poor bioavailability, high-dose requirements, adverse side effect, low therapeutic index and non-specific targeting, showing potential in clinical settings [27-29]. The success of drug-loaded nano-delivery system mainly relies on typical changes in the pharmacokinetic profile of API, such as the area under the plasma concentration-time curve (AUC), the elimination half-life (t1/2), the volume of distribution (Vd), total body clearance (CL) and the maximum plasma drug concentration (Cmax). The European Medicine Agency pointed out that correctly identifying the parameters that define relevant physicochemical properties of micelles is critical to ensure its quality [30].
The important criteria for new nanomedicines approval are based on the better therapeutic effect and safety profile. Clinical trials include a stepwise assessment of the safety and efficacy, and are divided into three phases: Phase Ⅰ (mainly assesses safety, including dose-limiting toxicity (DLT), maximum tolerated dose (MTD) and pharmacokinetic profile), Phase Ⅱ (mainly evaluates efficacy, including the objective response rate, progression-free survival (PFS) and overall survival), and Phase Ⅲ (mainly evaluates safety, efficacy and dosage) [31]. Treatment will be discontinued when the disease progresses or unacceptable toxicities develop. This review aims to provide an up to date overview on the current development of PM and their clinical performance.
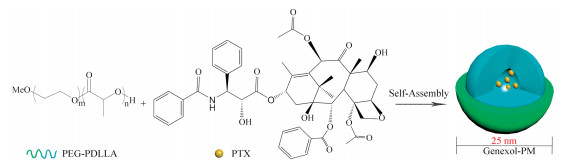
2. PM in clinical trials and on the market 2.1. Genexol-PM (anti-tumor drug)Paclitaxel (PTX, C47H51NO14, 853.92 g/mol) is the prototype of the taxane class of chemotherapeutics as part of the National Cancer Institute screen of plants and natural products with putative anticancer activity, which can inhibit microtubule depolymerization of free tubulins [32-34]. TaxolⓇ is a formulation of PTX with a 1:1 mixture of polyoxyethylene castor oil (Cremophor EL) and dehydrated alcohol, however, this non-ionic surfactant at the needed doses of the drug frequently produces acute hypersensitivity reactions [36]. Genexol-PM (Samyang Co., Seoul, Korea), marketed as Cynviloq or IG-001, is a PTX-loaded poly(ethylene glycol)2000-b-poly(D, L-lactide)1750 (PEG2000-b-PDLLA1750) micellar formulation with a 25 nm diameter, and that decreases the toxicity associated with Cremophor EL administration, as shown in Fig. 2 [33, 35, 36]. It is the first polymeric micellar product to the market, and Sorrento Therapeutics Inc. announced the acquisition of exclusive distribution rights from Samyang to Genexol-PM in the European Union and in the United States (after merging with IgDraSol Inc.). For constructing Genexol-PM, the block copolymer and drug were dissolved in acetonitrile, and stirred to evaporate the organic solvent. PTX-incorporated micelles were obtained by dissolving the resulting transparent gel matrix with preheated water.

|
Download:
|
| Fig. 2. Schematic diagram of the preparation of Genexol-PM. The micelle carrier of Genexol-PM consists of the block copolymer of PEG (molecular weight (MW) of about 2000 Da) and PDLLA (MW 1750 Da). | |
Genexol-PM has been approved for the treatment of breast cancer, non-small-cell lung cancer and ovarian cancer [38-40]. The expansion of Genexol-PM in other cancer clinical applications are undergoing clinical evaluation [40-42]. A Phase Ⅱ study of Genexol-PM in patients with locally advanced or metastatic pancreatic cancer showed that Genexol-PM at a dose of 300 mg/m2 every three weeks was well tolerated and common toxicities were similar to Taxol [42]. The overall responses rate was 6.7%, with 1 patient in complete response and 2 patients in partial responses, and the disease control rate was 60%. Median PFS and median overall survival were 2.8 months and 6.5 months, respectively. Grade 3 toxicities included neutropenia (40.0%), fatigue (17.8%), infection (13.3%), dehydration (13.3%), neuropathy (13.3%), and abdominal pain (11.1%). A Phase Ⅱ study of Genexol-PM (240 mg/m2) in patients with advanced urothelial cancer previously treated with Gemcitabine and Platinum combination chemotherapy (n = 37) showed Genexol-PM was well tolerated and had sufficient antitumor activity as second-line chemotherapy after Gemcitabine-Platinum failure in patients with urothelial cancer [41]. Of 34 evaluable patients, the overall responses rate was 21%, with 1 patient in complete response. Median PFS and median overall survival were 2.7 months and 6.5 months, respectively. Grade 3/4 non-hematologic toxicities included neutropenia (14.7%) and infection (5.9%). Only 1 patient occurred grade 3/4 hematologic toxicities.
A Phase Ⅱ study of Genexol-PM (230 mg/m2) in combination with Cisplatin (70 mg/m2) every three weeks for six cycles in 42 patients with unresectable thymoma (n = 14) or thymic carcinoma (n = 28) showed that Genexol-PM combined with Cisplatin had comparable efficacy to that of anthracycline-based regimens and was well tolerated [40]. For 40 evaluable patients, the objective responses were 62.5% (46% for thymoma (n = 13) and 70% for thymic carcinoma (n = 27)). The median PFS was 9.8 months (11.4 months for thymoma and 8.1 months for thymic carcinoma) and the two-year overall survival was 77.9% for thymoma and 65.9% for thymic carcinoma. Myelosuppression with grade 3/4 neutropenia was observed in 26% patients (n = 11). Thymoma has been reported to have a better prognosis and a more sensitive response to chemotherapy than thymic carcinoma [43, 44]. However, we found a higher response rate for thymic carcinoma than for thymoma, and a possible explanation is that a higher proportion of patients with thymoma were exposed to previous chemotherapy compared to thymic carcinoma (31% vs. 15%). In another Phase Ⅱ study of Genexol-PM, patients with advanced non-small cell lung cancer (n = 43) received Genexol-PM (230 mg/m2) on day 1 and Gemcitabine (1000 mg/m2) on day 1 and day 8 every three weeks for six cycles [45]. The overall response rate, median PFS and median overall survival were 46.5%, 4.0 months and 14.8 months, respectively. The most common grade 3/4 toxicities included neutropenia (16%) and pneumonia (12%). Notably, two patients died of pneumonia and dyspnea. Genexol-PM combined with Gemcitabine demonstrated the favorable antitumor activity in non-small cell lung cancer, but the safety remains to be further studied. Nevertheless, the results of these clinical trials showed an encouraging response rate, which led to proceeding with Phase Ⅲ and Phase Ⅳ clinical evaluations.
Specific clinical information of Genexol-PM from ClinicalTrials.gov database is shown in Table 1 [46].
|
|
Table 1 Clinical information of Genexol-PM (size: 25 nm, block copolymer: PEG-PDLLA, API: PTX, DL: -) from ClinicalTrials.gov database. |

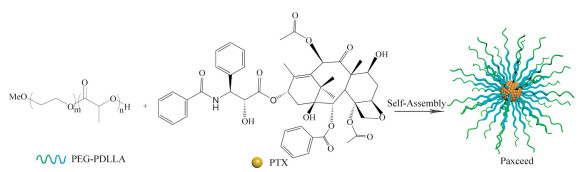
As a microtubule stabilizer, PTX can disrupt the normal dynamic reorganization of the microtubule network necessary for mitosis and cell proliferation. In addition, PTX can arrest the cell cycle at G2/M phase, and inhibit tumor-associated angiogenesis and tumor cell proliferation. Given the aggressive proliferation of synovial cells and pannus formation in rheumatoid arthritis (RA), PTX therapy for RA treatment is not surprising [47-49]. Paxceed (Angiotech Pharmaceuticals, Inc., Vancouver, BC, Canada) is a PTX-loaded PEG2000-b-PDLLA1333 micellar formulation with a PTX loading of 25% (w/w), as shown in Fig. 3 [50, 51]. The dose of Paxceed used for RA is much lower than that used for cancer. For constructing Paxceed, PTX and the copolymer were dissolved in acetonitrile followed by evaporation of the solvent. Micellar solution was obtained by dissolving the resulting matrix with preheated water.

|
Download:
|
| Fig. 3. Schematic diagram of the preparation of Paxceed. The micelle carrier of Paxceed consists of the block copolymer of PEG (MW 2000 Da) and PDLLA (MW 1333 Da). | |
A Phase Ⅱ pilot Study of Paxceed in patients with severe psoriasis (n = 12) was conducted in Maryland [52]. At the stage Ⅰ, Paxceed (75 mg/m2) was intravenously infused every four weeks for six cycles. Due to a decrease in efficacy three to four weeks after each infusion, at the stage Ⅱ, the adjusted dose of 37.5 mg/m2 was intravenously infused every two weeks for three cycles followed by 50 mg/m2 of Paxceed for 6 cycles. In the stage Ⅰ, all 5 patients improved (mean = 59.7% decrease in psoriasis area and severity index, range 40.3%-79.2%). Four patients who completed stage Ⅱ (n = 7) improved (mean = 45.9% decrease in psoriasis area and severity index, range 14.6%-79.1%). Paxceed was well tolerated throughout stage Ⅰ and Ⅱ. Phase Ⅲ clinical trials of Paxceed are currently in progress, but no new was reported since 2004.
Specific clinical information of Paxceed from ClinicalTrials.gov database is shown in Table 2 [53].
|
|
Table 2 Clinical information of Paxceed (size: -, block copolymer: PEG-PDLLA, API: PTX, DL: 25%) from ClinicalTrials.gov database. |

NK105 (NanoCarrier/Nippon Kayaku, Japan) is a PTX-loaded PEG12000-b-poly (4-phenyl-1-butanoate-L-aspartamide)8000 (PEG12000-b-PPBA8000) micellar formulation with a 85 nm diameter and a PTX loading of 23% (w/w), as shown in Fig. 4 [54, 55]. The half of carboxylic groups of the polyaspartate block (P(Asp)) were esterified with 4-phenyl-1-butanol to increase the hydrophobicity of the core. For constructing NK105, the block copolymer and PTX were dissolved in dichloromethane and dispersed in water as an emulsion. After evaporation of dichloromethane, the block copolymers self-assembled into core-shell PM incorporating PTX [56, 57].

|
Download:
|
| Fig. 4. Schematic diagram of the preparation of NK105. The micelle carrier of NK105 consists of the block copolymer of PEG (MW 12,000 Da) and P(Asp) modified with 4-phenyl-1-butanoate (MW 8000 Da). | |
In a Phase Ⅱ study, NK105 (150 mg/m2) was administered intravenously every three weeks in patients with advanced gastric cancer after failure of first-line chemotherapy without anti-allergic premedication (n = 56) [58]. The overall response rate was 25.0%, with 2 patients in complete responses and 12 patients in partial responses. 30% patients experienced stable disease for several months. The median PFS, median overall survival and time to treatment failure were 3.0 months, 14.4 months and 2.8 months, respectively. Drug related main toxicities were neutropenia (64.9%), leukopenia (17.5%), lymphopenia (8.8%), neuropathysensory (1.8%), fatigue (3.5%) and stomatitis (1.8%). NK105 at a dose of 150 mg/m2 exhibited a 9-fold larger plasma AUC, a 26-fold lower CL and a 10-fold lower Vd compared with conventional PTX at a dose of 210 mg/m2.
Two independent Phase Ⅰ studies of NK105 with once every three weeks schedule (at doses ranging from 10-180 mg/m2) or weekly schedule (at doses ranging from 50-100 mg/m2) were conducted, which showed that weekly NK105 (80 mg/m2) was well tolerance and presented a desirable antitumor activity profile [59, 60]. A Phase Ⅲ non-inferiority study comparing NK105 and PTX in patients with metastatic or recurrent breast cancer (n = 436) was started in 2012 and completed in July 2019 [61]. Although the recommended weekly dose of NK105 was 80 mg/m2, neutropenia was frequently observed in the subsequent expansion stages. The dose had to be reduced to be 65 mg/m2 based on the results from the Phase Ⅰ study [59]. NK105 (65 mg/m2) and PTX (80 mg/m2) was administered intravenously once weekly for three consecutive weeks. 3.3% patients (n = 214) for NK105 and 10.8% patients (n = 213) for PTX discontinued treatment due to adverse effect (neutropenia and leukopenia). The incidence of peripheral sensory neuropathy (PSN) for NK105 and PTX were 1.4% vs. 7.5%, respectively. The median PFS, the median overall survival and overall response rates for NK105 and PTX were 8.4 months vs. 8.5 months, 31.2 months vs. 36.2 months and 31.6% vs. 39.0%, respectively. Unexpectedly, the median PFS of PTX was longer than NK105, which may be due to a lower dose intensity of NK105. In 2016, Nippon Kayaku Co., Ltd. announced that "the primary endpoint of the study, progression free survival, did not meet the prespecified statistical criteria", but NK105 was well tolerated and had a better PSN profile than PTX.
Specific clinical information of NK105 from ClinicalTrials.gov database is shown in Table 3 [62].
|
|
Table 3 Clinical information of NK105 (size: 85 nm, block copolymer: PEG-PPBA, API: PTX, DL: 23%) from ClinicalTrials.gov database. |

Paccal Vet (Oasmia Pharmaceutical) is a novel nanoparticle formulation of PTX in combination with a novel excipient composed of a surfactant-based derivative of retinoic acid (XR-17: N-all-trans retinoyl cysteine methyl ester sodium salt and N-13-cis retinoyl cysteine methyl ester sodium salt) with a 20-40 nm diameter, as shown in Fig. 5 [63, 64].

|
Download:
|
| Fig. 5. Schematic diagram of the preparation of Paccal Vet. The micelle carrier of Paccal Vet consists of XR-17 (N-all-trans retinoyl cysteine methyl ester sodium salt and N-13-cis retinoyl cysteine methyl ester sodium salt). | |
A multi-center European, open label, single-arm study of Paccal Vet in 29 dogs with grade Ⅱ or Ⅲ mast cell tumors showed the clinically significant biological activity of Paccal Vet against mast cell tumors [65]. Paccal Vet was administered infused at doses ranging from 135 mg/m2 to 150 mg/m2 every three weeks for three cycles. Complete or partial responses were observed in 59% dogs. The median time to response and median PFS were 15 days and 247 days, respectively. Grade 3/4 neutropenia and grade 1/2 leukopenia were observed in the most dogs. 13 dogs were discontinued the study due to disease progression, with 9 dogs that were euthanased and 1 dog that died. A Phase Ⅲ study of Paccal Vet vs. lomustine in 252 dogs with grade Ⅱ or Ⅲ mast cell tumors showed Paccal Vet had clinically safe and effective [66]. Overall response rates for Paccal Vet and lomustine were 7% vs. 1%, respectively. The majority adverse events of Paccal Vet were transient and clinically manageable. 27 dogs (33%) treating with lomustine developed hepatopathy and were discontinued compared with 3 dogs (2%) treating with Paccal Vet.
Paccal Vet has been granted MUMS (Minor Uses/Minor Species, similar to orphan drug designation) status and was approval by the FDA Center for Veterinary Medicine for use in dogs with resectable and nonresectable squamous cell carcinoma and nonresectable stage Ⅲ, IV and V mammary carcinoma [32]. Federal law prohibits extralabel use of conditionally approved drugs, meaning that Paccal Vet can only be utilized for aboved indications.
The trials of Paccal Vet are not listed in the ClinicalTrials.gov database and the clinical information of Paccal Vet from published literatures is shown in Table 4 [65, 66].
|
|
Table 4 Clinical information of Paccal Vet (size: 20-40 nm, block copolymer: XR-17, API: PTX, DL: -) from published literatures. |

Docetaxel (C43H53NO14, 807.9 g/mol) is a semi-synthetic taxoid derived from the European yew and the first promising PTX analog. Docetaxel is about twice as potent as PTX as an inhibitor of microtubule depolymerisation, which was used widely as an antimicrotubule agent in breast, lung, ovarian and gastric cancers [67-70]. The clinical docetaxel formulation Taxotere is composed of polysorbate 80 and ethanol, associated to several adverse effects, including hypersensitivity reactions, hemolysis and peripheral neuropathy [71, 72]. BIND-014 (BIND Biosciences, Cambridge, MA, USA) is a novel targeted nanoparticles with a 100 nm diameter, which is composed of PEG-PLA decorated with small-molecule prostate-specific membrane antigen substrate analog inhibitor (S, S-2-[3-[5-amino-1-carboxypentyl]-ureido]-pentanedioic acid, ACUPA), encapsulating docetaxel, as shown in Fig. 6 [73, 74].

|
Download:
|
| Fig. 6. Schematic diagram of the preparation of BIND-014. The micelle carrier of BIND-014 consists of the block copolymer of PEG-PLA and ACUPA-PEG-PLA. | |
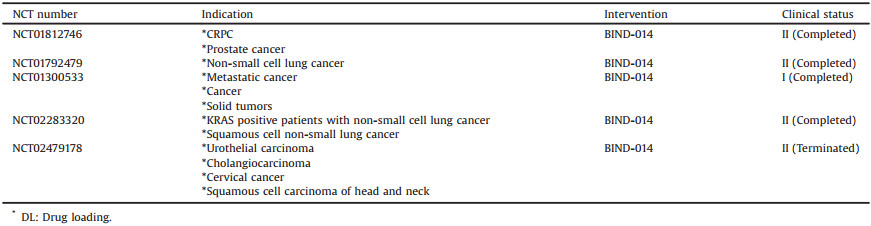
A Phase Ⅰ study of BIND-014 in patients with advanced or metastatic tumors demonstrated BIND-014 was well-tolerated with predictable and manageable toxicity [73]. The recommended Phase Ⅱ dose for BIND-014 is 60 mg/m2 every three weeks or 40 mg/m2 weekly. A multicenter open-label, Phase Ⅱ study of BIND-014 in combination with prednisone in patients with metastatic castration-resistant prostate cancer (n = 42) was conducted in 2013 [75]. BIND-014 (60 mg/m2) was intravenously administered every three weeks along with prednisone (5 mg/m2) twice daily. Drug-related adverse effect included fatigue (69%), nausea (55%), neuropathy (33%), and neutropenic fever (2%). The overall responses rate was 32%, with 1 patient in complete response, 5 patients in partial responses and 9 patients in stable diseases, and the median PFS was 9.9 months. In another Phase Ⅱ study, BIND-014 (60 mg/m2) was intravenously administered every three weeks as the second-line therapy in 40 patients with Stage Ⅲ/Ⅳ non-small cell lung cancer with characterized genomic status (EGFR mutation, ALK rearrangement, KRAS mutation). 63% disease control rate was observed in patients with KRAS mutations. The common adverse effects such as neutropenia, anemia and neuropathy were significantly reduced by BIND-014. Although BIND-014 was somewhat effective against lung cancer, it was disappointed in later clinical trials against cervical and head-and-neck cancers (data not published). In April 2016, the company announced that it would cut down its work with BIND-014, and explore new targets [76].
Specific clinical information of BIND-014 from ClinicalTrials.gov database is shown in Table 5 [77].
|
|
Table 5 Clinical information of BIND-014 (size: 100 nm, block copolymer: PEG-PLA & ACUPA-PEG-PLA, API: Docetaxel, DL: -) from ClinicalTrials.gov database. |
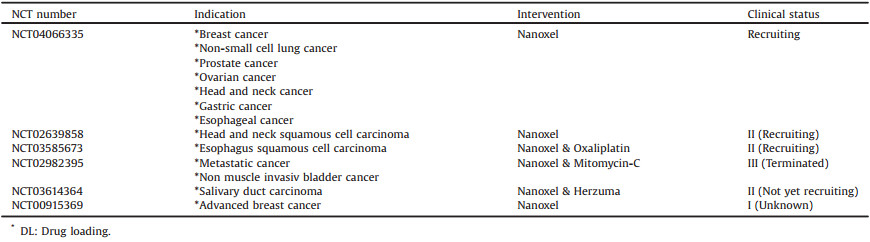
Nanoxel-PM (Samyang Co., Seoul, Korea), is a docetaxel-loaded PEG2000- PDLLA1765 micellar formulation with a 25 nm diameter, as shown in Fig. 7 [78, 79]. For constructing Nanoxel-PM, Docetaxel (4.0%, w/w) and the copolymer (76.0%, w/w) were dissolved in ethanol, and stirred to evaporate the organic solvent. Micellar solution was obtained by dissolving the resulting transparent gel matrix with preheated water. D-Mannitol (20.0%, w/w) aqueous solution as a cryoprotectant was added to the micellar solution, stirred for 20 min at room temperature.

|
Download:
|
| Fig. 7. Schematic diagram of the preparation of Nanoxel-PM. The micelle carrier of Nanoxel-PM consists of the block copolymer of PEG (MW 2000 Da) and PLA (MW 1765 Da). | |
The relative magnitude of AUCt and Cmax of Nanoxel-PM to those of TaxotereⓇ was 106.2% and 107.0% for mice, 98.8% and 91.0% for rats, and 88.0% and 82.7% for beagle dogs, respectively, which were within 100% ± 20% regarded as being bioequivalent [78]. Clinical studies had been registered in the ClinicalTrials.gov, but no results were published.
Specific clinical information of Nanoxel PM from ClinicalTrials.gov database is shown in Table 6 [80].
|
|
Table 6 Clinical information of Nanoxel-PM (size: 25 nm, block copolymer: PEG-PLA, API: Docetaxel, DL: -) from ClinicalTrials.gov database. |
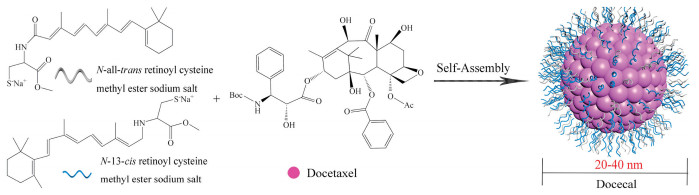
Docecal (Oasmia Pharmaceutical) is a next-generation nanoparticle formulation based on docetaxel, in combination with XR-17 with a carrier to API ratio of 2.25:1, as shown in Fig. 8.

|
Download:
|
| Fig. 8. Schematic diagram of the preparation of Docecal. The micelle carrier of Docecal consists of XR-17 (N-all-trans retinoyl cysteine methyl ester sodium salt and N-13-cis retinoyl cysteine methyl ester sodium salt). | |
Docecal was approved for clinical trials in 2015. Preclinical studies of Docecal demonstrated similar efficacy to TaxotereⓇ in inhibiting cell growth. The pharmacokinetic Phase Ⅰ study demonstrated that Docecal is bioequivalent with TaxotereⓇ (AUC0-∞ and Cmax) with regards to the total fraction of docetaxel in plasma. No unexpected adverse events were reported. In a Phase Ⅱ study, Docecal (100 mg/m2) without premedication or TaxotereⓇ (100 mg/m2) with standard mandated premedication was administered intravenously every three weeks for six cycles in 200 patients with metastatic breast cancer. Dexamethasone premedication was given at a dose of 16 mg/day (8 mg twice per day) for three days starting one day prior docetaxel administration. Drug related main toxicities were neutropenia (Docecal, 52% vs. TaxotereⓇ, 83%), leukopenia (Docecal, 15% vs. TaxotereⓇ, 27%) and febrile neutropenia (Docecal, 14% vs. TaxotereⓇ, 23%). The best overall objective tumor response (the results from main analysis) showed that non-inferiority with respect to efficacy could not be determined for Docecal. After finalization of the treatment for patients that completed all six cycles, additional statistical analyses with respect to efficacy showed Docecal met non-inferiority compared to TaxotereⓇ within the predefined limits. The efficacy as objective response rate is comparable at a later timepoint than defined in the main analysis.
The trials of Docecal are listed in the EU Clinical Trials Register is shown in Table 7 [81].
|
|
Table 7 Clinical information of Docecal (size: -, block copolymer: XR-17, API: Docetaxel, DL: -) from EU Clinical Trials Register. |

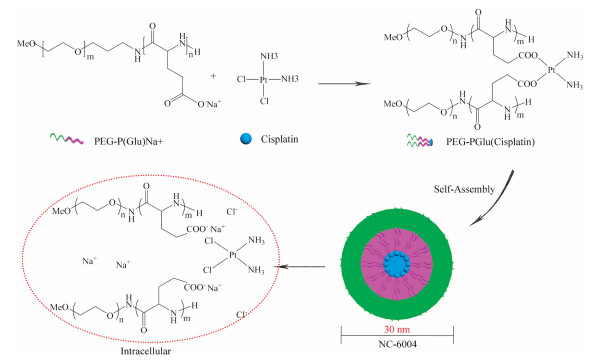
Cisplatin (Cl2H6N2Pt, 300.05 g/mol) is a platinum chelate complex and displays genotoxicity, activated intracellularly by the aquation of the two chloride leaving groups, and then covalently binds to DNA, forming DNA adducts [82]. Kataoka and colleagues developed the first PM loaded with Cisplatin, formed via polymer-metal complex formation between the carboxylic groups of PEG-b-p(α, β-aspartic acid) and cisplatin in water [83]. However, the rapid structural decay of the micelles led to high levels of accumulation of drugs in the liver and spleen, which resulted in similar antitumor efficacy as the free drug. To improve the stability of the PM and the drug release profile, NC-6004 (NanoCarrier, Japan/Orient EuroPharma, co-development, Fig. 9), a Cisplatin-loaded PEG12000-b-poly(L-glutamic acid)6000 (PEG-P(Glu)) micellar formulation with a 30 nm diameter and a Cisplatin loading of 39% (w/w) was developed. For constructing NC-6004, the sodium salt of PEG-P(Glu) and Cisplatin [(Glu) = 5 mmol/liter; (Cisplatin)/(Glu) = 1.0]) were dissolved in distilled water and reacted for 72 h to prepare Cisplatin-incorporating micelles [84-87]. In media containing chloride ions, Cisplatin is released from NC-6004 through exchange reaction between the carboxylic groups in P(Glu) and the chloride ions.

|
Download:
|
| Fig. 9. Schematic diagram of the preparation of NC-6004. The micelle carrier of NC-6004 consists of PEG12000-P(Glu)6000-Na+. | |
A Phase Ⅰ study of NC-6004 for patients with advanced solid tumors (n = 17) was conducted in UK, at doses ranging from 10 mg/m2 to 120 mg/m2 [88]. Renal impairment and hypersensitivity reactions were developed at 120 mg/m2 as MTD, and the recommended dose for Phase Ⅱ of NC-6004 was 90 mg/m2. The Cmax and AUC for NC-6004 at 120 mg/m2 were 34-fold smaller and 8.5-fold larger than those for Cisplatin, respectively. There were no patients in complete response or partial response, 41.2% patients in stable disease. The median PFS was 49 days and 82.4% patients died or had tumor progression. Another Phase Ⅰ study of NC-6004 in combination with gemcitabine for advanced solid tumors was conducted in Japan (n = 12) [89]. For the first treatment cycle (day 21/cycle) NC-6004 alone was administered at doses ranging from 60 mg/m2 to 90 mg/m2. From the second to eighth cycles, patients received NC-6004 (on day 1/cycle) in combination with 1000 mg/m2 of gemcitabine (on day 8/cycle). The most common drug-related adverse events were neutrophil decrease (66.7%) and white blood cell count decrease (41.7%). The MTD was determined to be 90 mg/m2 and the recommended dose for Phase Ⅱ was 60 mg/m2.
A Phase Ⅰ/Ⅱ trial of NC-6004 plus gemcitabine for metastatic pancreatic cancer was conducted in Taiwan and Singapore (n = 22) [90]. NC-6004 at doses ranging from 60 mg/m2 to 180 mg/m2 (on day 1/cycle) and 1250 mg/m2 of gemcitabine (on day 1 and day 8/cycle) was administered. Grade 3/4 leukopenia occurred in 68% patients and grade 3/4 thrombocytopenia occurred in 59% patients. Of 20 evaluable patients, tumor shrinkage occurred in 55%, partial responses in 15%, and stable disease in 70%. No complete response was observed. Since 2014, NC-6004 in combination with gemcitabine vs. gemcitabine alone has proceeded into Phase Ⅲ study in patients with locally advanced or metastatic pancreatic cancer, but the results have not been published yet.
Specific clinical information of NC-6004 from ClinicalTrials.gov database is shown in Table 8 [91].
|
|
Table 8 Clinical information of NC-6004 (size: 30 nm, block copolymer: PEG-P(Glu)Na, API: Cisplatin, DL: 39%) from ClinicalTrials.gov database. |

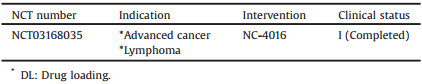
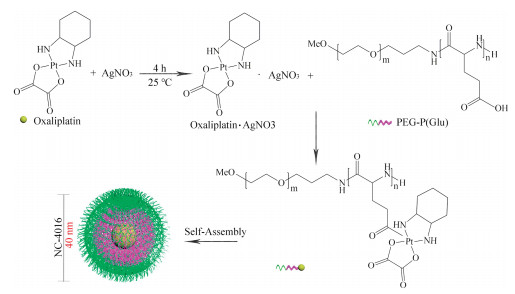
Although NC-6004 showed good stability and efficient drug disposal, cisplatin administration is associated with acute toxicity and acquired resistance. Oxaliplatin (C8H14N2O4Pt, 397.29 g/mol), as the third-generation platinum drug, is a water-soluble derivative of the failed drug tetraplatin, which is different from Cisplatin and Carboplatin in that it presents free amino groups linked to platinum and has lower toxicity and tumor resistance [92-95]. NC-4016 (NanoCarrier Co., Ltd., Japan) is an Oxaliplatin-loaded PEG12000-P(Glu)6000 micellar formulation with a 40 nm diameter and an Oxaliplatin loading of 32% (w/w), as shown in Fig. 10. For constructing NC-4016, Oxaliplatin first mixed with silver nitrate ([AgNO3]/[Oxaliplatin] = 1) in distilled water to form the aqueous complex (25 ℃, 24 h), and then is separated from the precipitated silver chloride (AgCl) by centrifugation (3000 rpm, 10 min) and filtration (0.22 μm). After that, methoxy PEG-P(Glu) is added to the solution of Oxaliplatin aqueous complex ([Glu] = 5 mmol/liter; [Oxaliplatin]/[Glu] = 1.0) and reacted 5 days to prepare Oxaliplatin-incorporating micelles [93, 96]. In aqueous medium, NC-4016 is formed by the coordinate complex formation between platinum in Oxaliplatin and carboxylic groups in P(Glu).

|
Download:
|
| Fig. 10. Schematic diagram of the preparation of NC-4016. The micelle carrier of NC-4016 consists of the block copolymer of PEG (MW 12,000 Da) and P(Glu) (MW 6000 Da). | |
Since 2000, Oxaliplatin has been extensively studied in combination with many chemotherapy drugs, such as Gemcitabine and Oxaliplatin in combination with Bevacizumab for advanced hepatocellular carcinoma [97], Capecitabine and Oxaliplatin compared with Fluorouracil/folinic acid and Oxaliplatin for metastatic colorectal cancer [98, 99], Oxaliplatin combined with 5-Fluorouracil (5-FU) or Gemcitabine for advanced pancreatic carcinoma [100-102], Oxaliplatin, Fluorouracil and Leucovorin for unresectable liver-only metastases from colorectal cancer [103], Oxaliplatin combined with 5-FU for esophageal cancer [104], Cetuximab combined with Oxaliplatin for metastatic gastric cancer [105], Bevacizumab, Oxaliplatin, and Capecitabine with radiation therapy for rectal cancer, showing an encouraging safety profile and response rates in different cancers.
A Phase Ⅰ dose-escalation and pharmacokinetic study of NC-4016 in patients with advanced solid tumors or lymphoma (n = 34) has been completed in 2017, at the University of Texas MD Anderson Cancer Center (Houston, Texas, United State). NC-4016 was intravenously infused at doses ranging from 15 mg/m2 to 80 mg/m2 every three weeks, but no results have been published so far.
Specific clinical information of NC-4016 from ClinicalTrials.gov database is shown in Table 9 [106].
|
|
Table 9 Clinical information of NC-4016 (size: 40 nm, block copolymer: PEG-P(Glu), API: Oxaliplatin, DL: 32%) from ClinicalTrials.gov database. |
Doxorubicin (Dox, C27H29NO11, 543.52 g/mol), known as AdriamycinⓇ or RubexⓇ, is a powerful anthracycline antibiotic isolated from a mutated strain of Streptomyces peucetius, which can mostly impair DNA replication along with RNA transcription by interaction with topoisomerase Ⅱ (Topo Ⅱ) [107-111]. Dox can be reduced intracellularly into a bioactive metabolite-doxorubicinol (Fig. 11) [112, 113]. NK911 (Nippon Kayaku, Japan), is a Dox-loaded PEG5000-poly(aspartic acid)4000 block copolymer conjugated with Dox (PEG-P(Asp-Dox)) micellar formulation with a 40 nm diameter, as shown in Fig. 11. To increase the hydrophobicity of the hydrophobic block and increase the compatibility and affinity between the core and the physically loaded Dox, Dox was partially conjugated (ca. 45%) to the carboxylic groups of the P(Asp) block via a hydrolytically stable amide bond. For constructing NK911, the block copolymer and drug were dissolved in the aqueous medium and drug was incorporated inside the core of the formed micelles [114]. It was the physically encapsulated Dox rather than the conjugated Dox that exerted antitumor activity.

|
Download:
|
| Fig. 11. (A) Schematic diagram of the preparation of NK911. The micelle carrier of NK911 consists of the block copolymer of PEG (MW 5000 Da) and conjugated Dox-P(Asp) (about 30 units). (B) A Dox's bioactive metabolite-Doxorubicinol. | |
In the Phase Ⅰ study, NK911 was administered infused at doses ranging from 6 mg/m2 to 67 mg/m2 every three weeks in 23 patients with metastatic or recurrent solid tumors refractory to conventional chemotherapy [115]. DLT was neutropenia observed at a dose of 67 mg/m2 (grade 4 neutropenia lasting more than 5 days), and this dosage level was determined to be the MTD. The recommended Phase Ⅱ dose of NK911 was 50 mg/m2 every three weeks. At 50 mg/m2, the plasma AUC of NK911 (3.2 μgh/mL) was 2-fold higher than that of free Dox (1.6 μgh/mL). Based on the good tolerability, preliminary signs of efficacy, and the lack of response associated with infusion, the decision was made to proceed with clinical development against metastatic pancreatic cancer. However, no news has been reported since 2004.
The trials of NK911 are not listed in the ClinicalTrials.gov database and the clinical information of NK911 from published literatures is shown in Table 10 [115].
|
|
Table 10 Clinical information of NK911 (size: 40 nm, block copolymer: PEG-P(Asp-Dox), API: Dox, DL: -) from published literatures. |

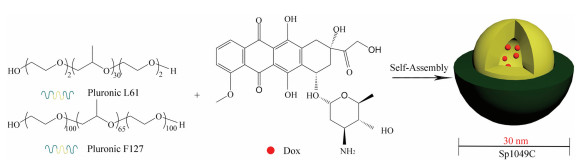
Sp1049C (Supratek Pharma, Inc., Montreal, Canada) is a micellar formulation composed of Dox physically loaded in a blend of two micellar forming non-ionic Pluronic block copolymers (Pluronic F127 and Pluronic L61 at a ratio of 1:8 (w/w)) with a 30 nm diameter and a Dox loading of 8.2% (w/w), as shown in Fig. 12. Pluronic F127 is used to stabilize the micelles, and Pluronic L61 is used for chemosensitization since it can inhibit the efflux transporter P-gp [116].

|
Download:
|
| Fig. 12. Schematic diagram of the preparation of Sp1049C. The micelle carrier of Sp1049C consists of Pluronic F127 and Pluronic L61. | |
In a Phase Ⅰ clinical trial, Sp1049C was administered infused at doses ranging from 5 mg/m2 to 90 mg/m2 every three weeks for at least six cycles in patients with advanced solid tumors (n = 26) [117]. The pharmacokinetic profile was linear (R2 = 0.71, Cmax and AUC0-∞ increases proportionally with doses) and the t1/2 obtained from Sp1049C (48.8 h) was longer than that of conventional Dox (30 h). The MTD was 70 mg/m2 and is recommended for Phase Ⅱ trials. The DLT was myelosuppression at 90 mg/m2 and non-hematological toxicity included alopecia, stomatitis and transient lethargy. Of 21 evaluable patients, no patients had a partial or complete response and 30.8% patients had stable disease with a median time to progression of 17.5 weeks. Sp1049C was granted orphan drug designation by FDA for therapy of esophageal carcinoma (in 2005) and gastric cancer (in 2008). A Phase Ⅱ clinical trial of Sp1049C (75 mg/m2) in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction (n = 21) was conducted in 2002, and showed a notable single-agent activity and an acceptable safety profile [118]. Of 19 evaluable patients, 9 patients had a partial response and 8 patients had stable disease. The objective response rate, median overall survival and PFS were 47%, 10 months and 6.6 months, respectively. Neutropenia was the predominant haematological toxicity as DLT. Although Sp1049C had reached Phase Ⅲ trial in patients with gastrointestinal cancer, no results have been reported since 2008.
The trials of Sp1049C are not listed in the ClinicalTrials.gov database, and the clinical information of Sp1049C from published literatures is shown in Table 11 [119].
|
|
Table 11 Clinical information of Sp1049C (size: 30 nm, block copolymer: Pluronic F127 & Pluronic L61, API: Dox, DL: -) from published literatures. |

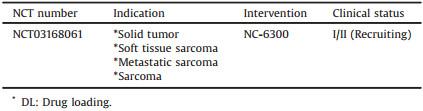
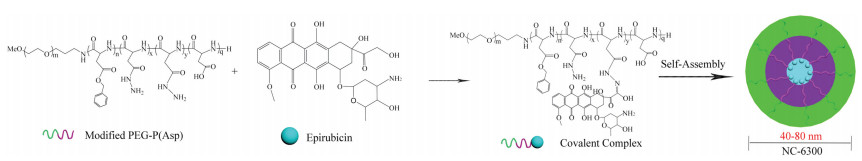
Epirubicin (C27H29NO11, 543.52 g/mol), is the 4′-Epirubicinmer of the anthracycline antibiotic Dox, and the cardiotoxicity of Epirubicin is ~66% of that of Dox [119-122]. NC-6300 (NanoCarrier Co., Ltd, Japan), known as K-912, comprises Epirubicin covalently bound to the PEG12000-polyaspartate (PEG12000-b-P(Asp)) block copolymer via an acid-labile hydrazone bond with a 40-80 nm diameter, as shown in Fig. 13 [123]. The P(Asp) is partially modified with benzyl groups to stabilize the micellar structure, and the number of Asp residues is 40 in which approximately 20 benzyl groups and eight molecules of Epirubicin are conjugated [124]. NC-6300 is pH-responsive, around 80% of Epirubicin was released at pH 3.0 within 1 h, whereas only about 20% of Epirubicin was released at pH 7.0 or pH 7.4 within 48 h.

|
Download:
|
| Fig. 13. Schematic diagram of the preparation of NC-6300. The micelle carrier of NC-6300 consists of the block copolymer of PEG (MW 12,000 Da) and P(Asp) partially modified with benzyl groups. The number of Asp residue is 40 (the sum of n, o, p and q = 40). | |
In Japan, a Phase Ⅰ clinical study of NC-6300 in 19 patients with advanced or recurrent solid tumors was conducted, showing NC-6300 was well tolerated and less toxicity than conventional Epirubicin formulation [123]. NC-6300 was administered at doses ranging from 15 mg/m2 to 225 mg/m2 every three weeks. The pharmacokinetic profile was linear (Cmax and AUC0-∞ increases proportionally with doses) and the Vd was approximately equal to the blood volume indicating minimal distribution. There was no significant cardiac related adverse event in patients at 15-225 mg/m2, even at 900 mg/m2. One patient with breast cancer had partial response at 100 mg/m2 and 10 patients had stable disease. The MTD and the recommended dose for Phase Ⅱ studies were determined to be 170 mg/m2. The Phase Ⅱ clinical study of NC-6300 is required to further define its antitumor activity and tolerability in patients with soft tissue sarcoma.
Specific clinical information of NC-6300 from ClinicalTrials.gov database is shown in Table 12 [125].
|
|
Table 12 Clinical information of NC-6300 (size: 40-80 nm, block copolymer: modified PEG-P(Asp), API: Epirubicin, DL: -) from ClinicalTrials.gov database. |
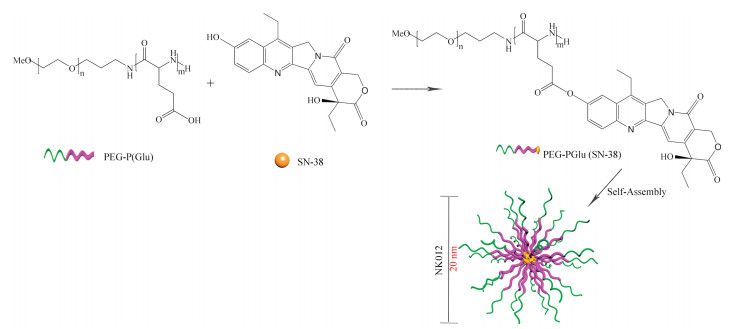
SN-38 (C22H20N2O5, 392.4 g/mol) is a biological active metabolite of irinotecan hydrochloride (CPT-11), which impedes DNA replication and transcription by interfering with the topoisomerase I (Topo I), approved by the FDA for the treatment of recurrent metastatic colorectal cancer [126, 127]. Although CPT-11 is converted to SN-38 in liver and tumor, the metabolic rate is lower (< 10%), and the conversion of CPT-11 to SN-38 depends on the genetic variation of the carboxylesterase activity between individuals. Therefore, further effective use of SN-38 may have tremendous advantages in cancer treatment [128]. SN-38 can be metabolized by UGT1A1 enzymes, and polymorphism of the UGT1A1 gene is associated with the incidence of severe neutropenia [129, 130]. Notably, at physiological pH, the lactone SN-38 with anticancer activity is partially converted into the inactive carboxylate [131]. NK012 (Nippon Kayaku, Co.), is a SN-38-loaded PEG12000-b-P(Glu)7000 micellar formulation with a 20 nm diameter and a SN-38 loading of 20% (w/w), as shown in Fig. 14 [130, 132]. The covalent of SN-38 was introduced into the P(Glu) segment by the condensation reaction between the carboxylic acid on P(Glu) and the phenol on SN-38 at 26 ℃ NK012 was constructed with self-assembling PEG12000-PGlu(SN-38) in an aqueous milieu.

|
Download:
|
| Fig. 14. Schematic diagram of the preparation of NK012. The micelle carrier of NK012 consists of the block copolymer of PEG (MW 12,000 Da) and P(Glu) (MW 7000 Da). | |
A Phase Ⅰ clinical trials in 38 patients with solid tumors (UGT1A1 *28 allele genotype of wt/wt and wt/*28), NK012 was intravenously infused at doses ranging from 9 mg/m2 to 37 mg/m2 every three or four weeks [133]. A higher plasma AUC of NK012 and a longer t1/2 value of polymer-unbound SN-38 at a dose of 28 mg/m2 (287 μgh/mL & 282 h) were observed compared to that of CPT-11 at a dose of 250 mg/m2 (27.86 μg h/mL & 13.9 h). Of 37 evaluable patients, 6 patients had partial responses, 18 patients had stable diseases and 12 patients had disease progression. The recommended Phase Ⅱ does of NK012 was 28 mg/m2 every four weeks. In Japan, a Phase Ⅰ clinical trials of NK012 determined the recommended dose for Phase Ⅱ trial was 28 mg/m2 and the DLT was neutropenia, in which patients were not homozygous or heterozygous for UGT1A1 *28 or UGT1A1 *6 [134]. Subsequently, a Phase Ⅱ trial of NK012 in unresectable, recurrent or metastatic colorectal cancer patients previously receiving Oxaliplatin-based chemotherapy was conducted [135]. Group A (53 patients with UGT1A1 genotype -/-, *6/-, or *28/- for evaluating the primary efficacy) and group B (5 patients with UGT1A1 genotype *6/*28 or *6/*6 for evaluating the reference) were administrated intravenously at the initial doses of 28 and 18 mg/m2 every three weeks. The disease control rate, the median PFS and overall survival were 56%, 3.30 months and 15.03 months, respectively. The overall response rate was 3.8%, which was similar to the irinotecan (4.2%) reported in the Phase Ⅲ EPIC trial. Based on the incidence and severity of DLT (grade ≥ 3 neutropenia), the initial dose of NK012 (28 mg/m2) may be excessive for patients. The optimal dose of NK012 needs further study to improve the efficacy and safety. In the USA, two Phase Ⅱ clinical trials of NK012 were underway in patients with relapse small cell lung cancer and triple negative breast cancer.
NK012 was employed in combination with different conventional drugs to further improve the efficacy of the monotherapy regimen, such as combined with 5-FU for colorectal cancer and combined with cisplatin for small cell lung cancer [136, 137].
Specific clinical information of NK012 from ClinicalTrials.gov database is shown in Table 13 [138].
|
|
Table 13 Clinical information of NK012 (size: 20 nm, block copolymer: PEG-P(Glu), API: SN-38, DL: 20%) from ClinicalTrials.gov database. |

Data from clinical studies suggested the potential and prospect of self-assembling PM as an alternative and efficacious drug delivery vehicle. PM can change the pharmacokinetics of encapsulated/conjugated drugs (a decrease in the Vd and CL, and an increase in the AUC and t1/2), resulting in a decreased toxicity compared to the conventional formulations (non-PM) allowing the use of higher drug doses in a clinic. In our review, four micellar formulations incorporating PTX (Genexol-PM, NK105, Paccal Vet and Paxceed) without Cremophor EL, two micellar formulations incorporating docetaxel (BIND-014, Nanoxel-PM and Docecal), two micellar formulations incorporating Dox (NK911 and Sp1049C), and one micellar formulation incorporating cisplatin, oxaliplatin, epirubicin or SN-38 (NC-6004, NC-4016, NC-6300 or NK012) are currently under clinical evaluation for different indications, mostly showing the positive clinical results. Additionally, the major clinical trials of PMs incorporating PTX and its derivative-docetaxel have basically completed Phase Ⅱ clinical trials (Table 14), showing a good trend of development. However, NK105 and BIND-014 were declared a failure due to a lack of efficacy, which could be explained by the heterogeneity among patients in clinical trials, appropriate regulatory guidelines and instability under biological environment. This efficacy may be further improved if the dose and administration schedule are modified. The proprietary of information results in many unpublished clinical results, including the Phase Ⅲ clinical trials of Paxceed, Sp1049C and NC-6004 combined with gemcitabine, and the clinical trials of Nanoxel PM.
|
|
Table 14 The major clinical trials of PTX and its derivatives-docetaxel. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Due to the hydrophobic character of many anticancer drugs, the development of PM-based therapies focusing on the treatment of cancer has attracted widespread attention (as video). Several formulations have been investigated in clinical studies, and Genexol-PM has been approved by the FDA. PM can be preferentially accumulated in tumors utilizing the leaky nature of the tumor vasculature and impaired lymphatic drainage, however, this mechanism has recently been questioned [139-142]. Warren C. W. Chan team demonstrated that the dominant mechanism by which nanoparticles entry into tumors is active transcytosis process rather than EPR [139]. BIND-014 is a unique multifunctional nanoparticles complex with targeting ligand investigated in clinical trials. Modulation of cell specificity by introduction of targeting ligands has the potential for overcoming biological barriers. Also, depending on the pharmacodynamic profile of the incorporated drug, triggered release by exogenous light/sound, or endogenous microenvironmental cues (pH or enzymatic activity), could be envisioned to further improve the therapeutic efficacy [143]. Such a multifunctional system is bound to increase the complexity in the development process.
Cancer is a complex disease involving multiple pathways and has led to great interest in combination treatments [144]. Inhibiting singleness of therapeutic target may be insufficient to achieve tumor recession, and the synergistic effect of multiple pathways may provide the chance of eliminating the cancer. Furthermore, combination therapies usually achieve therapeutic synergy or a medicinal effect that is greater than the sum of each drug treatment alone (1 + 1 > 2). Improved survival and better response rates in clinical practice has further demonstrated the benefit of combination therapies. However, the effective administration of multiple drugs with the optimal dose ratio is complicated by dissimilar pharmacokinetics and distribution due to different metabolic rates within body. The novel 'two in one' approach that carries multiple therapeutics agents in one nanoparticle is under clinical investigation, such as a liposomal nanoparticle containing CPX-1 and paclitaxel/tanespimycin [145, 146]. A long-term use of chemotherapy is often hampered by the development of drug resistance, which is often attributed to gene mutations, gene amplification or epigenetic changes that affect the uptake, metabolism or export of drugs from single cells. Another important cause for anticancer drug resistance is the limited ability of drugs to penetrate tumor tissue and to reach all tumor cells at potentially lethal concentrations. Additionally, the heterogeneity of the tumor microenvironment leads to significant gradients in the rate of cell proliferation and the regions of hypoxia and acidity, all of which can affect the sensitivity of tumor cells to drug therapy [147148]. Adequate and rational combination therapy, especially the combination of chemotherapy and immunotherapy, can delay or prevent the emergence of drug-resistance mutants and increase the rates of clinical cures. In recent years, chemoimmunotherapy, combining chemotherapeutics and immunotherapeutic, has emerged as a promising approach for cancer treatment, with the advantages of cooperating two kinds of treatment mechanism and enhancing therapeutic effect [149]. Some chemotherapy drugs, such as anthracycline, not only can induce cancer cell death, but also can stimulate antitumor immunity by immunogenic cell death (ICD) [150]. Although Dox can stimulate antitumor immunity by inducing ICD, the immunogenicity of tumor cells killed was weak [151, 152]. Further investigation of the immunogenic potential of other drugs, either alone or in combination, is warranted. In addition, new nanomaterials with immunomodulator function or enhancing the function of immunomodulator are also being continuously explored in research.
The first generation of PMs (such as PEG-PLA) aimed at solubilizing hydrophobic agents, however, PMs are administered intravenously and accompanied by micelle dilution in the bloodstream (the concentration will drop below CMC), resulting in (partial) micelle dissolution. Different strategies, including chemical crosslinking of PMs core and drug, covalent coupling of the drug to the polymer chains (such as NC-6004, NC-6300 and NK012), and hydrophobic interactions between the drug and the hydrophobic block of the copolymer, have been developed to achieve improved retention and stability of drug-loaded PMs [87]. In addition, the binding of the block copolymers to blood components (such as albumin and apolipoproteins) can also initiate micelle dissociation and premature drug loss [153]. Therefore, the growing number of cell-derived nanocarriers provides a glimpse of the drug delivery potential behind biological functionalization.
In general, there has little consideration on the relationship between the disease, patient pathophysiology and physicochemical properties of nanomedicine, so that knowledge regarding the in vivo fate and biophysical and chemical interaction of nanomedicines remains limited [5]. In silico models with better explanatory and predictive power has been used to investigate the influence of a range of parameters in biologically realistic scenarios (realistic models of the tumors), which not only can screen out the optimum nanoparticles based on patient-specific data but also minimize costly trial-and-error design methods [154]. Notably, this presents a new opportunity to allow us to investigate the influence of a range of designs and understand the complex immune clearance mechanisms in biologically relevant scenarios.
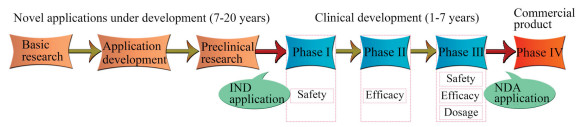
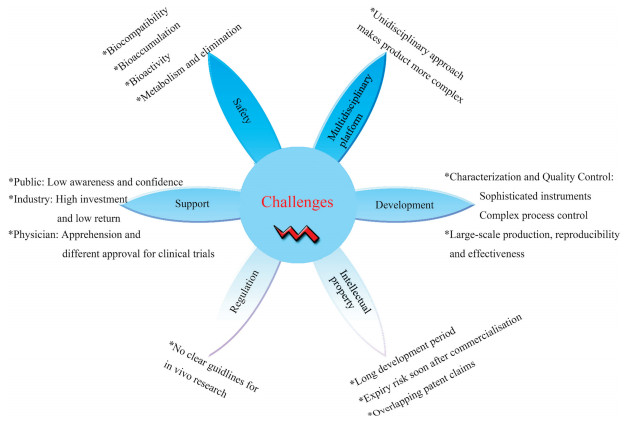
PM offers enormous advantages but is equally challenging. The current evaluation of safety, toxicity and compatibility of nanomedicines is based on FDA existing regulations for conventional drugs. Nanomedicines begin clinical trials to investigate their safety and efficacy in humans after gaining the investigational new drug (IND) application. After approval for clinical trials, the FDA revokes the IND and approves the new drug application (NDA). The general process of nanomedicines development is shown in Fig. 15 [155-157]. However, questions have been raised about the FDA's current approach for regulating nanomedicines, and critics oppose adapting and applying existing regulations [158, 159]. Therefore, there is an urgent need for a formal regulatory guideline specifically applicable to nanomedicines, which must be specific and sufficiently rigorous to ensure the safety and efficacy of nanoproducts [158, 159]. In addition to the inadequate regulatory guidelines, the clinical translation of nanomedicines still faces multiple challenges, such as reproducible and cost-effective manufacturing and scale-up; availability of characterization methods; safety concern; instability in biological environments; and insufficient understanding of patient disease heterogeneity [160-163]. Challenges to the clinical translation of nanomedicines are summarized in Fig. 16, which have to be addressed with ongoing advances in science and technology.

|
Download:
|
| Fig. 15. Schematic diagram of the general process of nanomedicines development. | |
{kind=link}

|
Download:
|
| Fig. 16. Challenges to the clinical translation of nanomedicines. | |
{kind=link}
Overall, nanotechnology has become closely related with cancer care today and has been applied in an evolutionary manner to improve the properties of cancer therapeutic. To achieve an efficient clinical translation, collaboration among academia, industry and regulatory bodies is required to ensure safe and effective nanomedicines.
4. OutlookPM-based drug delivery systems have a broad application prospect in clinical research. Despite all the formidable challenges, the prospect of this emerging field is still limitless. In the future, advances in this field will revolutionize the clinical landscape, improve our therapeutic and diagnostic facilities, and ultimately achieve the long-term success of tumor therapy.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsThis work was financially supported by Fundamental Research Funds for the Central Universities (No. 201964018), National Natural Science Foundation of China (No. 31872754), Qingdao Program for Original Innovation and Fundamental Research (No. 18-2-2-73-jch).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.029.
| [1] |
J.N. Meng, V. Agrahari, I. Youm, J. Neuroimmune Pharmacol. 12 (2016) 84-98. |
| [2] |
D. Peer, J.M. Karp, S. Hong, et al., Nat. Nanotechnol. 2 (2007) 751-760. DOI:10.1038/nnano.2007.387 |
| [3] |
A.C. Anselmo, S. Mitragotri, Bioeng. Transl. Med. 1 (2016) 10-29. DOI:10.1002/btm2.10003 |
| [4] |
C.I. Ochubiojo, M.E. Ibanga, S. Ifianyi, Nanotechnology in drug delivery, in: A. D. Sezer (Ed.), Recent Advances in Novel Drug Carrier Systems, Intech, Croatia, 2012.
|
| [5] |
V. Agrahari, V. Agrahari, Drug Discov. Today 23 (2018) 974-991. DOI:10.1016/j.drudis.2018.01.047 |
| [6] |
V. Sainz, J. Conniot, A.I. Matos, et al., Biochem. Biophys. Res. Commun. 468 (2015) 504-510. DOI:10.1016/j.bbrc.2015.08.023 |
| [7] |
S. Hassan, G. Prakash, A.B. Ozturk, et al., Nano Today 15 (2017) 91-106. DOI:10.1016/j.nantod.2017.06.008 |
| [8] |
A. Pandit, D.I. Zeugolis, Nanomedicine: N. B. M. 11 (2016) 985-987. DOI:10.2217/nnm.16.42 |
| [9] |
J.H. Xie, Y. Lu, B.Q. Yu, et al., Chin. Chem. Lett. 31 (2020) 1173-1177. DOI:10.1016/j.cclet.2019.10.030 |
| [10] |
P. Srikanth, R. Narayana, S.R. Wasim, et al., Adv. Pharm. 3 (2013) 51-58. |
| [11] |
Y. Wang, X.Q. Liang, R.S. Tong, et al., J. Biomed. Nanotechnol. 14 (2018) 1695-1704. DOI:10.1166/jbn.2018.2626 |
| [12] |
Q.Q. Hu, L. Bai, Z.J. Zhu, Chin. Chem. Lett. 31 (2020) 915-918. DOI:10.1016/j.cclet.2020.01.008 |
| [13] |
Y. Zhou, A.P. Fang, F.Z. Wang, et al., Chin. Chem. Lett. 31 (2020) 494-500. DOI:10.1016/j.cclet.2019.04.048 |
| [14] |
X. Zhang, X. Ren, J. Tang, et al., Drug Deliv. 27 (2020) 825-835. DOI:10.1080/10717544.2020.1770373 |
| [15] |
Y. Liang, Q.Q. Huo, W. Lu, et al., J. Biomed. Nanotechnol. 14 (2018) 1308-1316. DOI:10.1166/jbn.2018.2585 |
| [16] |
Y. Qu, B. Chu, X. Wei, et al., J. Control. Release 296 (2019) 93-106. DOI:10.1016/j.jconrel.2019.01.016 |
| [17] |
S.S. Kulthe, Y.M. Choudhari, N.N. Inamdar, et al., Des. Monomers Polym. 15 (2012) 465-521. DOI:10.1080/1385772X.2012.688328 |
| [18] |
M. Talelli, M. Barz, C.J.F. Rijcken, et al., Nano Today 10 (2015) 93-117. DOI:10.1016/j.nantod.2015.01.005 |
| [19] |
Y. Wang, X.X. Wang, J. Zhang, et al., Chin. Chem. Lett. 30 (2019) 885-888. DOI:10.1016/j.cclet.2019.02.018 |
| [20] |
C.L. Zhu, S. Gong, J.S. Ding, et al., Acta Pharm. Sin. B 9 (2019) 107-117. DOI:10.1016/j.apsb.2018.09.004 |
| [21] |
M. Cagel, F.C. Tesan, E. Bernabeu, et al., Eur. J. Pharm. Biopharm. 113 (2017) 211-228. DOI:10.1016/j.ejpb.2016.12.019 |
| [22] |
Z. Zhang, Q. Sun, J. Zhong, et al., J. Biomed. Nanotechnol. 10 (2014) 216-226. DOI:10.1166/jbn.2014.1729 |
| [23] |
K. Miyata, N. Nishiyama, K. Kataoka, Chem. Soc. Rev. 41 (2012) 2562-2574. DOI:10.1039/C1CS15258K |
| [24] |
N. Nishiyama, K. Kataoka, Adv. Polym. Sci. 193 (2015) 67-101. |
| [25] |
X. Xu, W. Ho, X. Zhang, et al., Trends Mol. Med. 21 (2015) 223-232. DOI:10.1016/j.molmed.2015.01.001 |
| [26] |
J.M. Caster, A.N. Patel, T. Zhang, A. Wang, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 9 (2017) 1-18. |
| [27] |
H. Havel, G. Finch, P. Strode, et al., AAPS J. 18 (2016) 1-6. DOI:10.1208/s12248-015-9820-y |
| [28] |
C.L. Ventola, Dermatol. Ther. 37 (2012) 631-639. |
| [29] |
D. Bobo, K.J. Robinson, J. Islam, et al., Pharm. Res. 33 (2016) 2373-2387. DOI:10.1007/s11095-016-1958-5 |
| [30] |
E.W. Party, C.F.R.F. Consultation, Stat. Med. 25 (2010) 1639-1645. |
| [31] |
A.J.M. Ferreri, T. Calimeri, G.M. Conte, et al., Blood 134 (2019) 252-262. |
| [32] |
C. Khanna, M. Rosenberg, D.M. Vail, J. Vet. Intern. Med. 29 (2015) 1006-1012. DOI:10.1111/jvim.12596 |
| [33] |
Z.Y. Fan, C. Chen, X.Y. Pang, et al., PLoS One 10 (2015) e0120129. DOI:10.1371/journal.pone.0120129 |
| [34] |
E. Miele, G.P. Spinelli, F. Tomao, Int. J. Nanomed. Nanosurg. 99 (2009) 99-105. |
| [35] |
C.P. Yang, S. Horwitz, Int. J. Mol. Sci. 18 (2017) 1733. DOI:10.3390/ijms18081733 |
| [36] |
S.C. Kim, D.W. Kim, Y.H. Shim, et al., J. Control. Release 72 (2001) 191-202. DOI:10.1016/S0168-3659(01)00275-9 |
| [37] |
K.S. Lee, H.C. Chung, S.A. Im, et al., Breast Cancer Res. Tr. 108 (2007) 217-222. |
| [38] |
D.W. Kim, S.Y. Kim, H.K. Kim, et al., Ann. Oncol. 18 (2007) 2009-2014. DOI:10.1093/annonc/mdm374 |
| [39] |
T.Y. Kim, J.Y. Chung, Clin. Cancer Res. 10 (2004) 3708-3716. DOI:10.1158/1078-0432.CCR-03-0655 |
| [40] |
H.S. Kim, J.Y. Lee, S.H. Lim, et al., J. Thorac. Oncol. 10 (2015) 762-768. DOI:10.1097/JTO.0000000000000489 |
| [41] |
J.L. Lee, J.H. Ahn, S.H. Park, et al., Invest. New Drugs 30 (2012) 1984-1990. DOI:10.1007/s10637-011-9757-7 |
| [42] |
M.W. Saif, N.A. Podoltsev, M.S. Rubin, et al., Cancer Invest. 28 (2010) 186-194. |
| [43] |
S. Margaritora, A. Cesario, G. Cusumano, et al., Ann. Thorac. Surg. 89 (2010) 245-252. DOI:10.1016/j.athoracsur.2009.08.074 |
| [44] |
G.L. Lemma, J.W. Lee, S.C. Aisner, et al., J. Clin. Oncol. 29 (2011) 2060-2065. DOI:10.1200/JCO.2010.32.9607 |
| [45] |
H.K. Ahn, S.J. Sym, et al., Cancer Chemoth. Pharm. 74 (2014) 277-282. DOI:10.1007/s00280-014-2498-5 |
| [46] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=GenexolPM&cntry=&state=&city=&dist=.
|
| [47] |
A. Saklani, S. Kutty, Drug Discov. Today 13 (2008) 161-171. DOI:10.1016/j.drudis.2007.10.010 |
| [48] |
Y. Zhao, Z.F. Chang, R. Li, et al., Am. J. Transl. Res. 8 (2016) 5044-5051. |
| [49] |
S. Jayashree, K. Nirekshana, G. Guha, D. Bhakta-Guha, Biomed. Pharmacother. 102 (2018) 894-911. DOI:10.1016/j.biopha.2018.03.123 |
| [50] |
X. Zhang, H.M. Burt, D.V. Hoff, et al., Cancer Chemoth. Pharm. 40 (1997) 81-86. DOI:10.1007/s002800050630 |
| [51] |
X. Yin, X. Cao, J. Li, et al., AAPS PharmSciTech 20 (2019) 629-638. |
| [52] |
A. Ehrlich, S. Booher, Y. Becerra, et al., J. Am. Acad. Dermatol. 50 (2004) 533-540. DOI:10.1016/j.jaad.2003.09.018 |
| [53] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=Paxceed&cntry=&state=&city=&dist=.
|
| [54] |
S. Emoto, H. Yamaguchi, J. Kishikawa, et al., Cancer Sci. 103 (2012) 1304-1310. DOI:10.1111/j.1349-7006.2012.02274.x |
| [55] |
I. Nakamura, E. Ichimura, R. Goda, et al., Int. J. Nanomedicine 12 (2017) 1293-1304. DOI:10.2147/IJN.S114356 |
| [56] |
T. Hamaguchi, Y. Matsumura, M. Suzuki, et al., Brit. J. Cancer 92 (2005) 1240-1246. DOI:10.1038/sj.bjc.6602479 |
| [57] |
T. Negishi, F. Koizumi, H. Uchino, et al., Brit. J. Cancer 95 (2006) 601-606. DOI:10.1038/sj.bjc.6603311 |
| [58] |
K. Kato, K. Chin, T. Yoshikawa, et al., Invest. New Drugs 30 (2011) 1621-1627. |
| [59] |
H. Mukai, K. Kato, T. Esaki, et al., Invest. New Drugs 34 (2016) 750-759. DOI:10.1007/s10637-016-0381-4 |
| [60] |
T. Hamaguchi, K. Kato, H. Yasui, et al., Br. J. Cancer 97 (2007) 170-176. DOI:10.1038/sj.bjc.6603855 |
| [61] |
Y. Fujiwara, H. Mukai, T. Saeki, et al., Brit. J. Cancer 120 (2019) 475-482. DOI:10.1038/s41416-019-0391-z |
| [62] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=NK105&cntry=&state=&city=&dist=.
|
| [63] |
H. von Euler, P. Rivera, H. Nyman, et al., Vet. Comp. Oncol. 11 (2013) 243-255. DOI:10.1111/j.1476-5829.2011.00314.x |
| [64] |
S. Hassan, S. Dhar, M. Sandström, et al., Cancer Chemoth. Pharm. 55 (2005) 47-54. DOI:10.1007/s00280-004-0855-5 |
| [65] |
P. Rivera, N. Åkerlund-Denneberg, K. Bergvall, et al., J. Small Anim. Pract. 54 (2013) 20-27. DOI:10.1111/j.1748-5827.2012.01304.x |
| [66] |
D.M. Vail, H. von Euler, A.W. Rusk, et al., J. Vet. Intern. Med. 26 (2012) 598-607. DOI:10.1111/j.1939-1676.2012.00897.x |
| [67] |
B.G.H. Choudhury, M. Pandey, et al., Int. J. Pharm. 529 (2018) 506-522. |
| [68] |
E.T. Oh, C.W. Kim, S.J. Kim, et al., Sci. Rep.-UK. 6 (2016) 1-37. DOI:10.1038/s41598-016-0001-8 |
| [69] |
J.J.M.A. Hendrikx, J.S. Lagas, J.Y. Song, et al., Int. J. Cancer 138 (2016) 758-769. DOI:10.1002/ijc.29812 |
| [70] |
S.Y. Song, K. Kim, S.Y. Jeong, Oncotarget 7 (2016) 77348-77357. DOI:10.18632/oncotarget.12668 |
| [71] |
Y.M. Yin, F.D. Cui, C.F. Mu, et al., J. Control. Release 140 (2009) 86-94. DOI:10.1016/j.jconrel.2009.08.015 |
| [72] |
G. Palma, C. Conte, A. Barbieri, et al., Int. J. Pharmaceut. 473 (2014) 55-63. DOI:10.1016/j.ijpharm.2014.06.058 |
| [73] |
D.D. Von Hoff, M.M. Mita, R.K. Ramanathan, et al., Clin. Cancer Res. 22 (2016) 3157-3163. DOI:10.1158/1078-0432.CCR-15-2548 |
| [74] |
J. Hrkach, D. Von Hoff, M.M. Ali, et al., Sci. Transl. Med. 4 (2012) 128-139. |
| [75] |
K.A. Autio, R. Dreicer, J. Anderson, et al., JAMA Oncol. 4 (2018) 1344-1351. DOI:10.1001/jamaoncol.2018.2168 |
| [76] |
H. Ledford, Biotechnology 533 (2016) 304-305. |
| [77] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=BIND-014&cntry=&state=&city=&dist=.
|
| [78] |
S.W. Lee, M.H. Yun, S.W. Jeong, et al., J. Control. Release 155 (2011) 262-271. DOI:10.1016/j.jconrel.2011.06.012 |
| [79] |
V.Q. Do, K.H. Park, J.M. Park, M.Y. Lee, Toxicol. Res. 35 (2019) 201-207. DOI:10.5487/TR.2019.35.2.201 |
| [80] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=Nanoxel&cntry=&state=&city=&dist=.
|
| [81] |
ClinicalTrials. gov database, (2020). https://www.Clinicaltrialsregister.eu/ctr-search/search?query=Docecal.
|
| [82] |
X. Duan, C. He, S.J. Kron, W. Lin, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 8 (2016) 776-791. DOI:10.1002/wnan.1390 |
| [83] |
M. Yokoyama, T. Okano, Y. Sakurai, et al., J. Control. Release 39 (1996) 351-356. DOI:10.1016/0168-3659(95)00165-4 |
| [84] |
Y. Mochida, H. Cabral, Y. Miura, et al., ACS Nano 8 (2014) 6724-6738. DOI:10.1021/nn500498t |
| [85] |
H. Uchino, Y. Matsumura, T. Negishi, et al., Brit. J. Cancer 93 (2005) 678-687. DOI:10.1038/sj.bjc.6602772 |
| [86] |
N. Nishiyama, S. Okazaki, H. Cabral, et al., Cancer Res. 63 (2013) 8977-8983. |
| [87] |
A. Varela-Moreira, Y. Shi, M.H.A.M. Fens, et al., Mater. Chem. Front. 1 (2017) 1485-1501. DOI:10.1039/C6QM00289G |
| [88] |
R. Plummer, R.H. Wilson, H. Calvert, et al., Brit. J. Cancer 104 (2011) 593-598. DOI:10.1038/bjc.2011.6 |
| [89] |
T. Doi, T. Hamaguchi, K. Shitara, et al., Cancer Chemoth. Pharm. 79 (2017) 569-578. DOI:10.1007/s00280-017-3254-4 |
| [90] |
V. Subbiah, J.E. Grilley-Olson, A.J. Combest, et al., Clin. Cancer Res. 24 (2018) 43-51. DOI:10.1158/1078-0432.CCR-17-1114 |
| [91] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=NC6004&cntry=&state=&city=&dist=.
|
| [92] |
J. Cassidy, J.L. Misset, Semin. Oncol. 29 (2002) 11-20. DOI:10.1053/sonc.2002.35524 |
| [93] |
H. Cabral, N. Nishiyama, S. Okazaki, et al., J. Control. Release 101 (2005) 223-232. DOI:10.1016/j.jconrel.2004.08.022 |
| [94] |
R.H. Wilson, T. Lehky, R.R. Thomas, et al., J. Clin. Oncol. 20 (2002) 1767-1774. DOI:10.1200/JCO.2002.07.056 |
| [95] |
L. Kelland, Nat. Rev. Cancer 7 (2007) 573-584. DOI:10.1038/nrc2167 |
| [96] |
H. Cabral, K. Kataoka, J. Control. Release 121 (2007) 146-155. DOI:10.1016/j.jconrel.2007.05.024 |
| [97] |
A.X. Zhu, L.S. Blaszkowsky, D.P. Ryan, et al., J. Clin. Oncol. 24 (2006) 1898-1903. DOI:10.1200/JCO.2005.04.9130 |
| [98] |
H.J. Schmoll, T. Cartwright, J. Tabernero, et al., J. Clin. Oncol. 26 (2007) 102-109. DOI:10.1200/JCO.2006.08.1075 |
| [99] |
J. Cassidy, S. Clarke, E. Díaz-Rubio, et al., J. Clin. Oncol. 26 (2008) 2006-2012. DOI:10.1200/JCO.2007.14.9898 |
| [100] |
M. Ducreux, E. Mitry, M. Ould-Kaci, et al., Ann. Oncol. 15 (2004) 467-473. DOI:10.1093/annonc/mdh098 |
| [101] |
C. Louvet, R. Labianca, P. Hammel, et al., J. Clin. Oncol. 23 (2005) 3509-3516. DOI:10.1200/JCO.2005.06.023 |
| [102] |
C. Louvet, T. André, G. Lledo, et al., J. Clin. Oncol. 20 (2002) 1512-1518. DOI:10.1200/JCO.2002.20.6.1512 |
| [103] |
S.R. Alberts, W.L. Horvath, W.C. Sternfeld, et al., J. Clin. Oncol. 23 (2005) 9243-9249. DOI:10.1200/JCO.2005.07.740 |
| [104] |
N.I. Khushalani, C.G. Leichman, G. Proulx, et al., J. Clin. Oncol. 20 (2002) 2844-2850. DOI:10.1200/JCO.2002.12.032 |
| [105] |
F. Lordick, B. Luber, S. Lorenzen, et al., Br. J. Cancer 102 (2010) 500-505. DOI:10.1038/sj.bjc.6605521 |
| [106] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=NC4016&cntry=&state=&city=&dist=.
|
| [107] |
C. Annabelle, C. Nina, T. Nouritza, et al., Oncotarget 9 (2018) 14539-14551. DOI:10.18632/oncotarget.24465 |
| [108] |
L.A.A. Gilliam, D.K. St. Clair, Antioxid. Redox Sign. 15 (2011) 2543-2563. DOI:10.1089/ars.2011.3965 |
| [109] |
G. Stefania, D.A. Antonella, B. Liberato, et al., Oxid. Med. Cell. Longev. 75 (2018) 1-15. |
| [110] |
C. Maximiliano, G. Estefania, B. Ezequiel, et al., Drug Discov. Today 22 (2017) 270-281. DOI:10.1016/j.drudis.2016.11.005 |
| [111] |
F. Arcamone, G. Cassinelli, G. Fantini, et al., Biotechnol. Bioeng. 67 (2000) 704-713. DOI:10.1002/(SICI)1097-0290(20000320)67:6<704::AID-BIT8>3.0.CO;2-L |
| [112] |
M.A. Mitry, J.G. Edwards, Int. J. Cardiol. Heart Vasc. 10 (2015) 17-24. |
| [113] |
D. Ren, M. Dalmau, A. Randall, et al., Adv. Funct. Mater. 22 (2012) 3170-3180. DOI:10.1002/adfm.201200052 |
| [114] |
T. Nakanishi, S. Fukushima, K. Okamoto, et al., J. Control. Release 74 (2001) 295-302. DOI:10.1016/S0168-3659(01)00341-8 |
| [115] |
Y. Matsumura, T. Hamaguchi, T. Ura, et al., Brit. J. Cancer 91 (2004) 1775-1781. DOI:10.1038/sj.bjc.6602204 |
| [116] |
A.V. Kabanov, E.V. Batrakova, V.Y. Alakhov, Adv. Drug Deliv. Rev. 54 (2002) 759-779. DOI:10.1016/S0169-409X(02)00047-9 |
| [117] |
S. Danson, D. Ferry, V. Alakhov, et al., Brit. J. Cancer 90 (2004) 2085-2091. DOI:10.1038/sj.bjc.6601856 |
| [118] |
J.W. Valle, A. Armstrong, C. Newman, et al., Invest. New Drugs 29 (2010) 1029-1037. |
| [119] |
A. Takahashi, Y. Yamamoto, M. Yasunaga, et al., Cancer Sci. 104 (2013) 920-925. DOI:10.1111/cas.12153 |
| [120] |
Y. Matsumura, J. Clin. Oncol. 44 (2014) 515-525. |
| [121] |
X.F. Wang, Z.F. Zhao, M.H. Chen, et al., Mol. Med. Rep. 12 (2015) 5917-5923. DOI:10.3892/mmr.2015.4220 |
| [122] |
L.S. Tarpgaard, C. Qvortrup, S.B. Nygard, et al., BMC Cancer 16 (2016) 91. DOI:10.1186/s12885-016-2124-5 |
| [123] |
H. Mukai, T. Kogawa, N. Matsubara, et al., Invest. New Drugs 35 (2017) 307-314. DOI:10.1007/s10637-016-0422-z |
| [124] |
M. Harada, I. Bobe, H. Saito, et al., Cancer Sci. 102 (2011) 192-199. DOI:10.1111/j.1349-7006.2010.01745.x |
| [125] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=+NC6300&cntry=&state=&city=&dist=.
|
| [126] |
C. Chaves, M. Taghi, M.C. Menet, et al., Pharmaceutics 12 (2020) 399-417. DOI:10.3390/pharmaceutics12050399 |
| [127] |
M. Sumitomo, F. Koizumi, T. Asano, et al., Cancer Res. 68 (2008) 1631-1635. DOI:10.1158/0008-5472.CAN-07-6532 |
| [128] |
Y. Matsumura, Adv. Drug Deliv. Rev. 63 (2011) 184-192. DOI:10.1016/j.addr.2010.05.008 |
| [129] |
F.F. Han, C.L. Guo, D. Yu, et al., Cancer Chemoth. Pharm. 73 (2014) 779-788. DOI:10.1007/s00280-014-2405-0 |
| [130] |
J. Gao, J. Zhou, Y. Li, et al., Medical Oncol. 30 (2013) 1-6. |
| [131] |
J.A. Zhang, T. Xuan, M. Parmar, et al., Int. J. Pharm. 270 (2004) 93-107. DOI:10.1016/j.ijpharm.2003.10.015 |
| [132] |
R. Zhang, R. Saito, Y. Mano, et al., Drug Deliv. 23 (2016) 2780-2786. DOI:10.3109/10717544.2015.1081994 |
| [133] |
H.A. Burris, J.R. Infante, F.A. Greco, et al., Cancer Chemother. Pharmacol. 77 (2016) 1079-1086. DOI:10.1007/s00280-016-2986-x |
| [134] |
T. Hamaguchi, T. Doi, T. Eguchi-Nakajima, et al., Clin. Cancer Res. 16 (2010) 5058-5066. DOI:10.1158/1078-0432.CCR-10-0387 |
| [135] |
T. Hamaguchi, A. Tsuji, K. Yamaguchi, et al., Cancer Chemother. Pharm. 82 (2018) 1021-1029. DOI:10.1007/s00280-018-3693-6 |
| [136] |
T. Nagano, M. Yasunaga, K. Goto, et al., Clin. Cancer Res. 15 (2009) 4348-4355. DOI:10.1158/1078-0432.CCR-08-3334 |
| [137] |
T.E. Nakajima, M. Yasunaga, Y. Kano, et al., Int. J. Cancer 122 (2008) 2148-2153. DOI:10.1002/ijc.23381 |
| [138] |
ClinicalTrials. gov database, (2020). https://www.clinicaltrials.gov/ct2/results?cond=&term=NK012&cntry=&state=&city=&dist=.
|
| [139] |
S. Sindhwani, A.M. Syed, J. Ngai, et al., Nat. Mater. 19 (2020) 566-575. DOI:10.1038/s41563-019-0566-2 |
| [140] |
S.M. Kim, P.H. Faix, J.E. Schnitzer, J. Control. Release 267 (2017) 15-30. DOI:10.1016/j.jconrel.2017.09.016 |
| [141] |
R. Daniel, T. Wei, Nat. Commun. 9 (2018) 1410. DOI:10.1038/s41467-018-03705-y |
| [142] |
A. Nel, E. Ruoslahti, H. Meng, ACS Nano 11 (2017) 9567-9569. DOI:10.1021/acsnano.7b07214 |
| [143] |
J. Liu, X.X. Ai, H.P. Zhang, et al., J. Biomed. Nanotechnol. 15 (2019) 373-381. DOI:10.1166/jbn.2019.2693 |
| [144] |
Y. Qu, B.Y. Chu, K. Shi, et al., J. Biomed. Nanotechnol. 13 (2017) 1598-1618. DOI:10.1166/jbn.2017.2475 |
| [145] |
L.M.K. Ma, S. Andrew, ACS Nano 7 (2013) 9518-9525. DOI:10.1021/nn405674m |
| [146] |
E.J. Feldman, J.E. Lancet, J.E. Kolitz, et al., J. Clin. Oncol. 29 (2011) 979-985. DOI:10.1200/JCO.2010.30.5961 |
| [147] |
O. Trédan, C.M. Galmarini, P. Krupa, et al., J. Natl. Cancer Inst. 99 (2007) 1441-1454. DOI:10.1093/jnci/djm135 |
| [148] |
N. Vasan, J. Baselga, D.M. Hyman, Nature 575 (2019) 299-309. DOI:10.1038/s41586-019-1730-1 |
| [149] |
W. Mu, Q. Chu, Y. Liu, N. Zhang, Nano-Micro Lett. 142 (2020). |
| [150] |
Z. Yu, J.F. Guo, M.G. Hu, et al., ACS Nano 14 (2020) 4816-4828. DOI:10.1021/acsnano.0c00708 |
| [151] |
N. Casares, M.O. Pequignot, A. Tesniere, et al., J. Exp. Med. 202 (2005) 1691-1701. DOI:10.1084/jem.20050915 |
| [152] |
J.M. Llovet, J. Zucman-Rossi, E. Pikarsky, et al., Nat. Rev. Dis. Primers 2 (2016) 16018. DOI:10.1038/nrdp.2016.18 |
| [153] |
S. Kim, Y. Shi, J.Y. Kim, et al., Expert Opin. Drug Del. 7 (2010) 49-62. DOI:10.1517/17425240903380446 |
| [154] |
N.R. Stillman, M. Kovacevic, I. Balaz, et al., NPJ Comput. Mater. 92 (2020). DOI:10.1038/s41524-020-00366-8 |
| [155] |
C. Fornaguera, M. García-Celma, J. Pers. Med. 7 (2017) 1-11. DOI:10.3390/jpm7010001 |
| [156] |
D.V. Bazile, J. Drug Deliv. Sci. Tec. 24 (2014) 12-21. DOI:10.1016/S1773-2247(14)50002-0 |
| [157] |
M.L. Etheridge, S.A. Campbell, A.G. Erdman, et al., Nanomedicine 9 (2013) 1-14. DOI:10.1016/j.nano.2012.05.013 |
| [158] |
J.H. Grossman, J.D. Clogston, AAPS J. 19 (2017) 92-102. DOI:10.1208/s12248-016-9980-4 |
| [159] |
S. Tinkle, S.E. McNeil, S. Mühlebach, et al., Ann. NY. Acad. Sci. 1313 (2014) 35-56. DOI:10.1111/nyas.12403 |
| [160] |
I.P. Kaur, V. Kakkar, P.K. Deol, et al., J. Control. Release 193 (2014) 51-62. DOI:10.1016/j.jconrel.2014.06.005 |
| [161] |
N. Desai, AAPS J. 14 (2012) 282-295. DOI:10.1208/s12248-012-9339-4 |
| [162] |
V. Agrahari, P. Hiremath, Nanomedicine 12 (2017) 819-823. DOI:10.2217/nnm-2017-0039 |
| [163] |
J.I. Hare, T. Lammers, M.B. Ashford, et al., Adv. Drug Deliv. Rev. 108 (2017) 25-38. DOI:10.1016/j.addr.2016.04.025 |